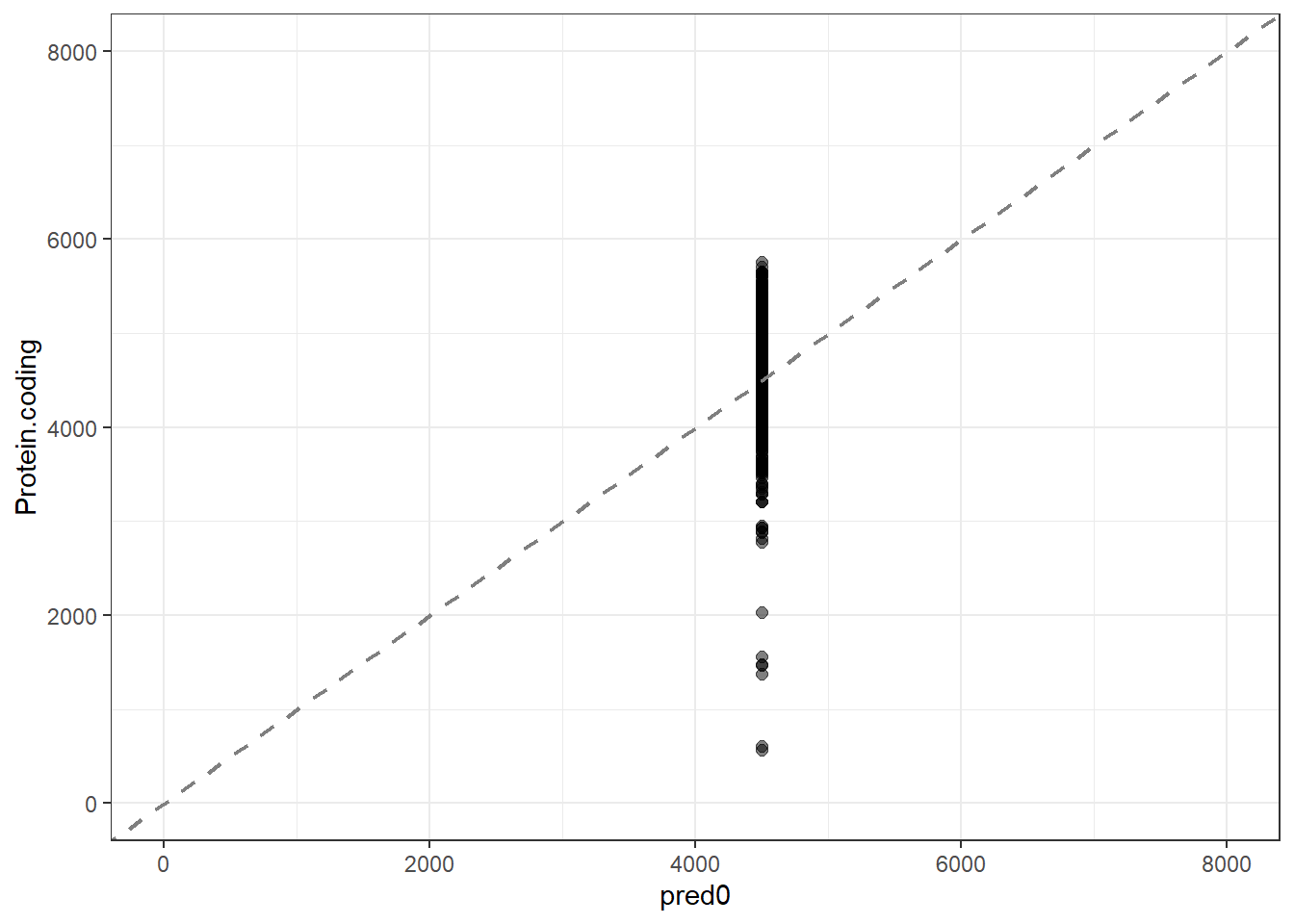
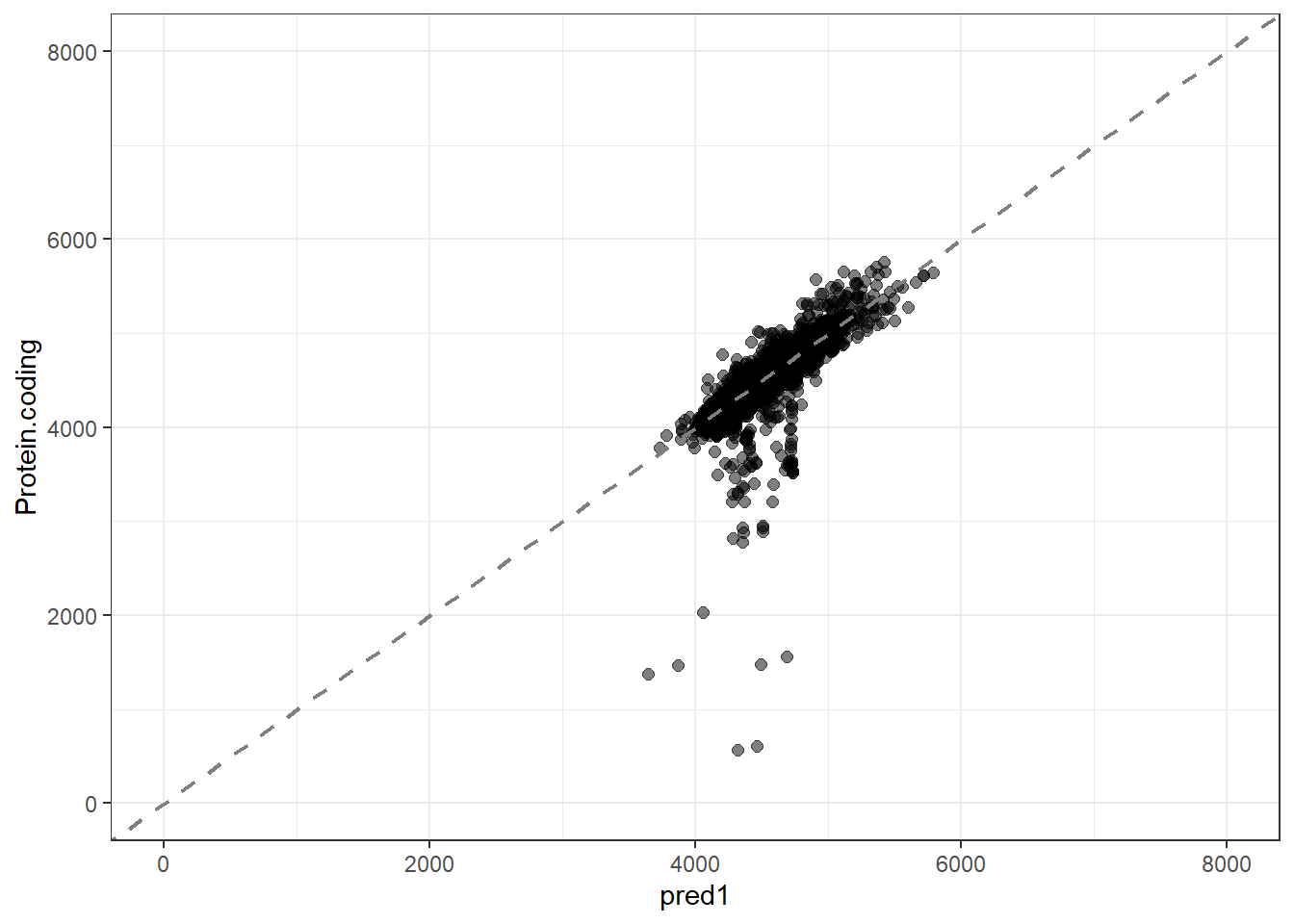
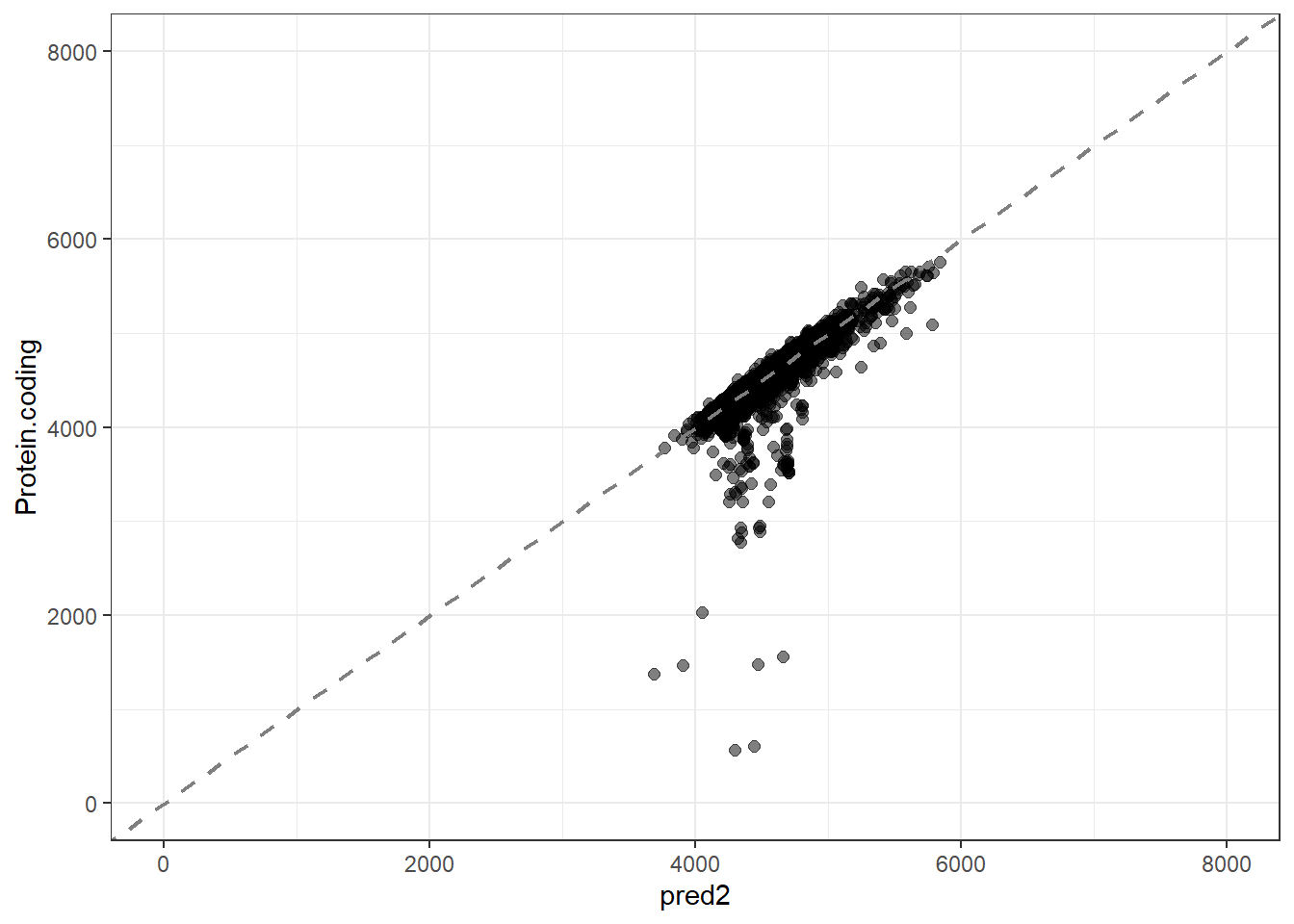
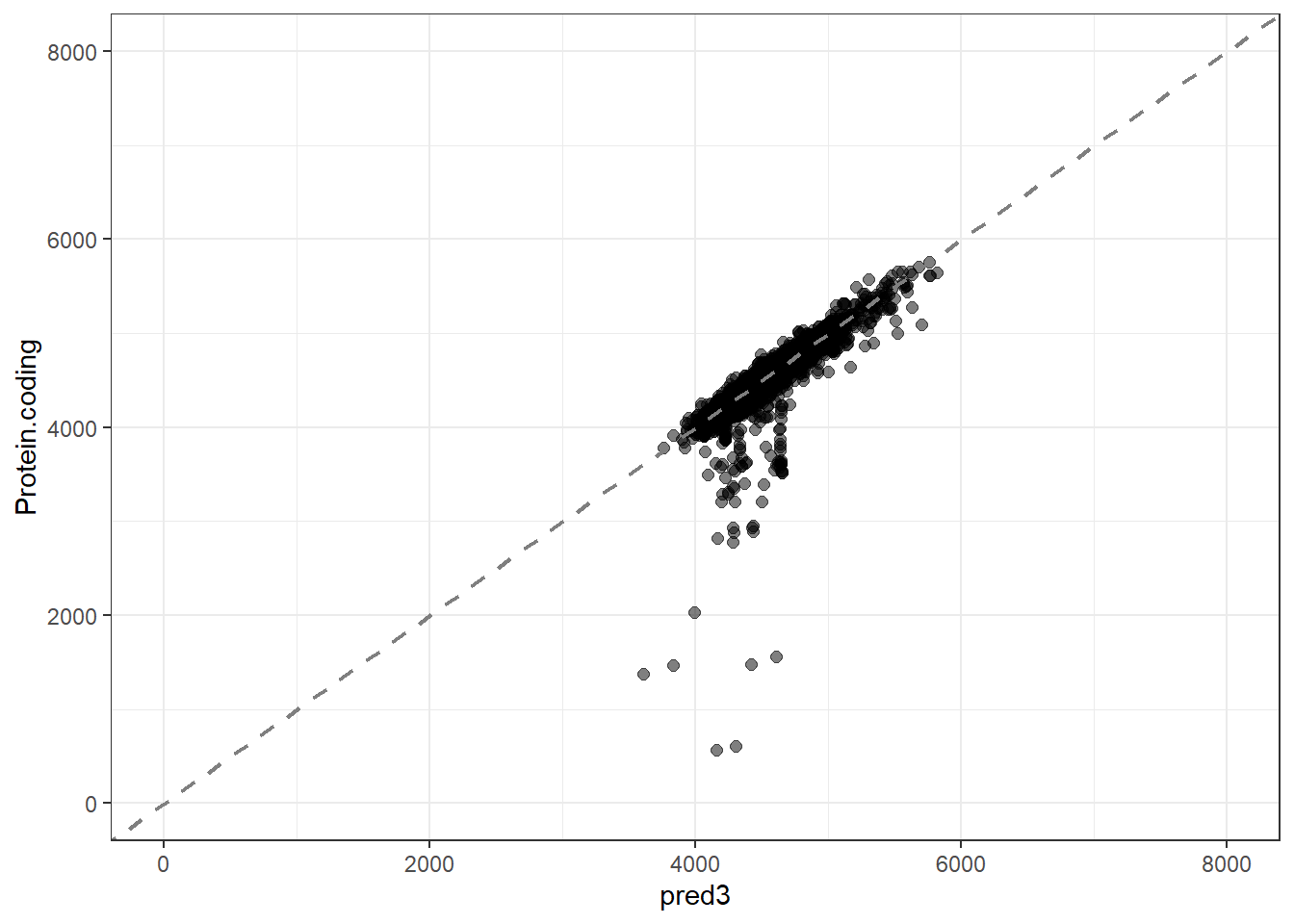
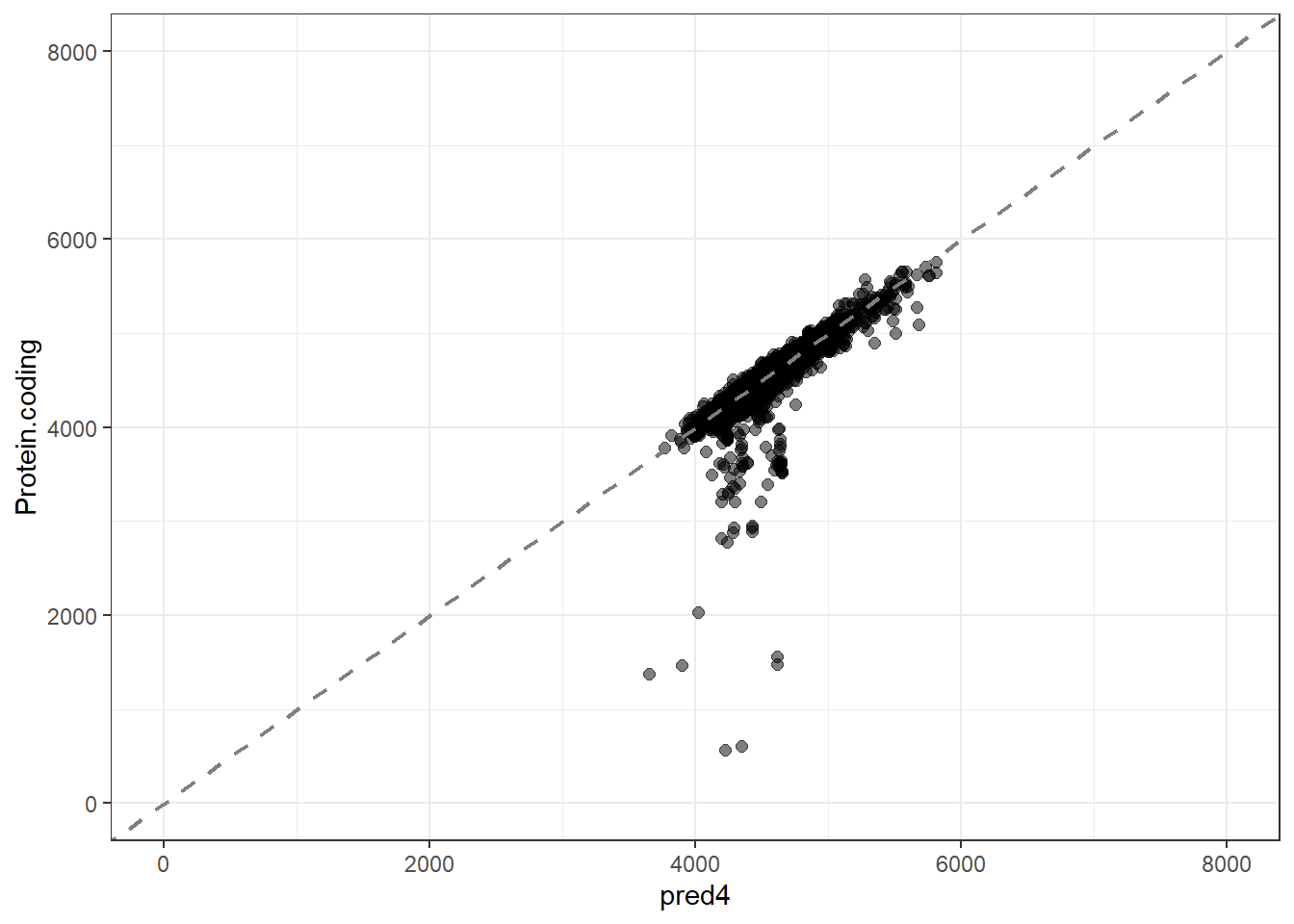
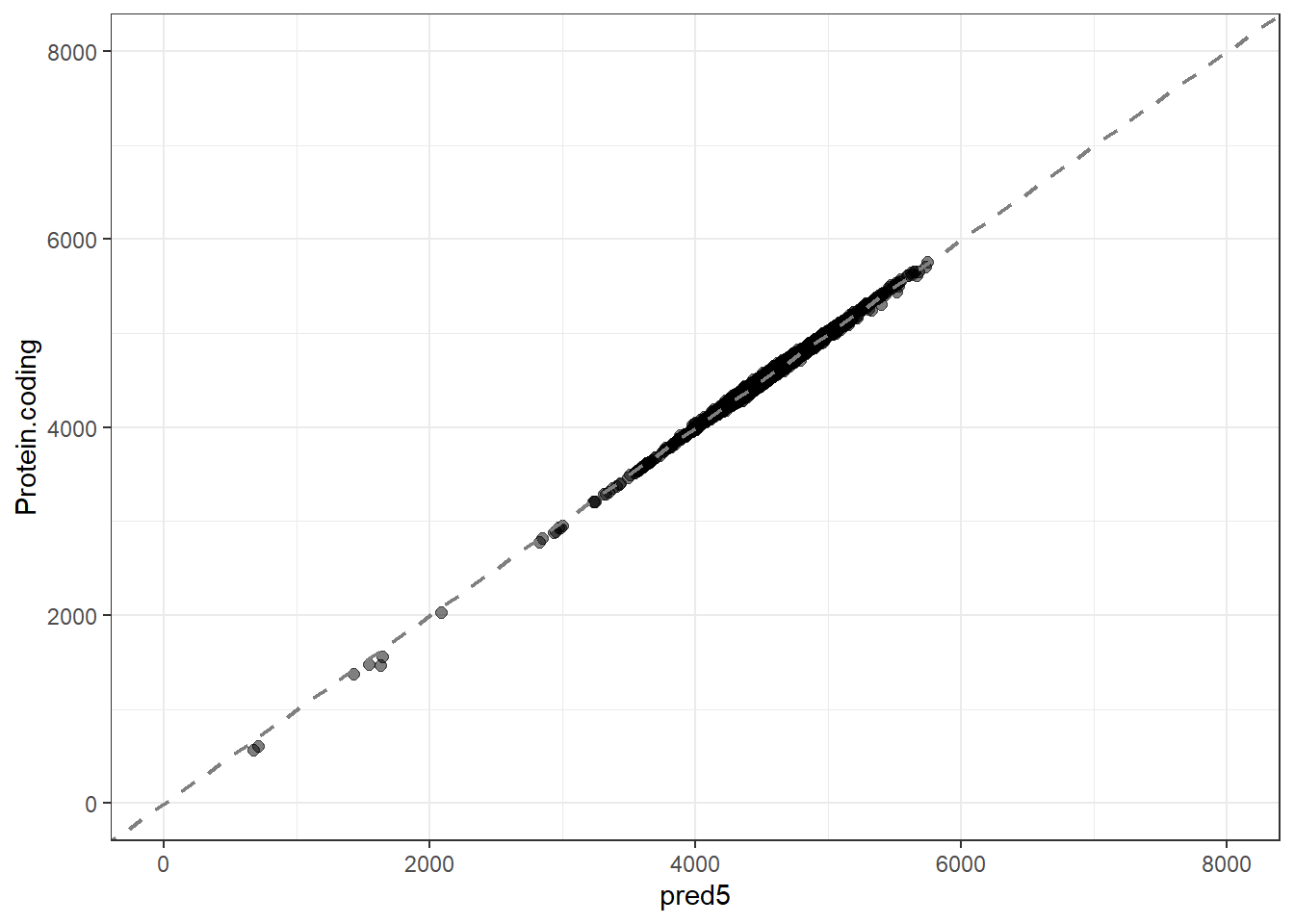
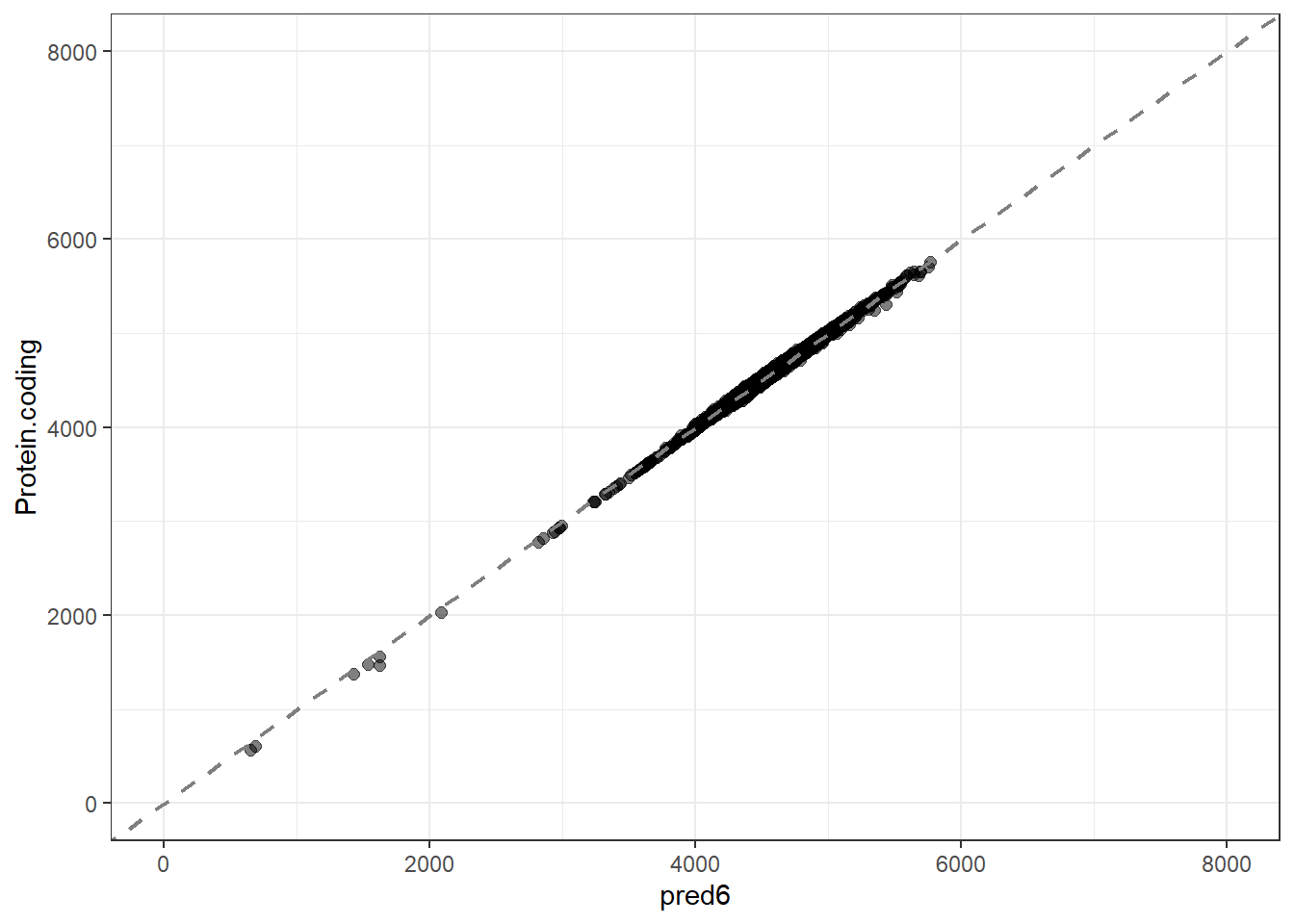
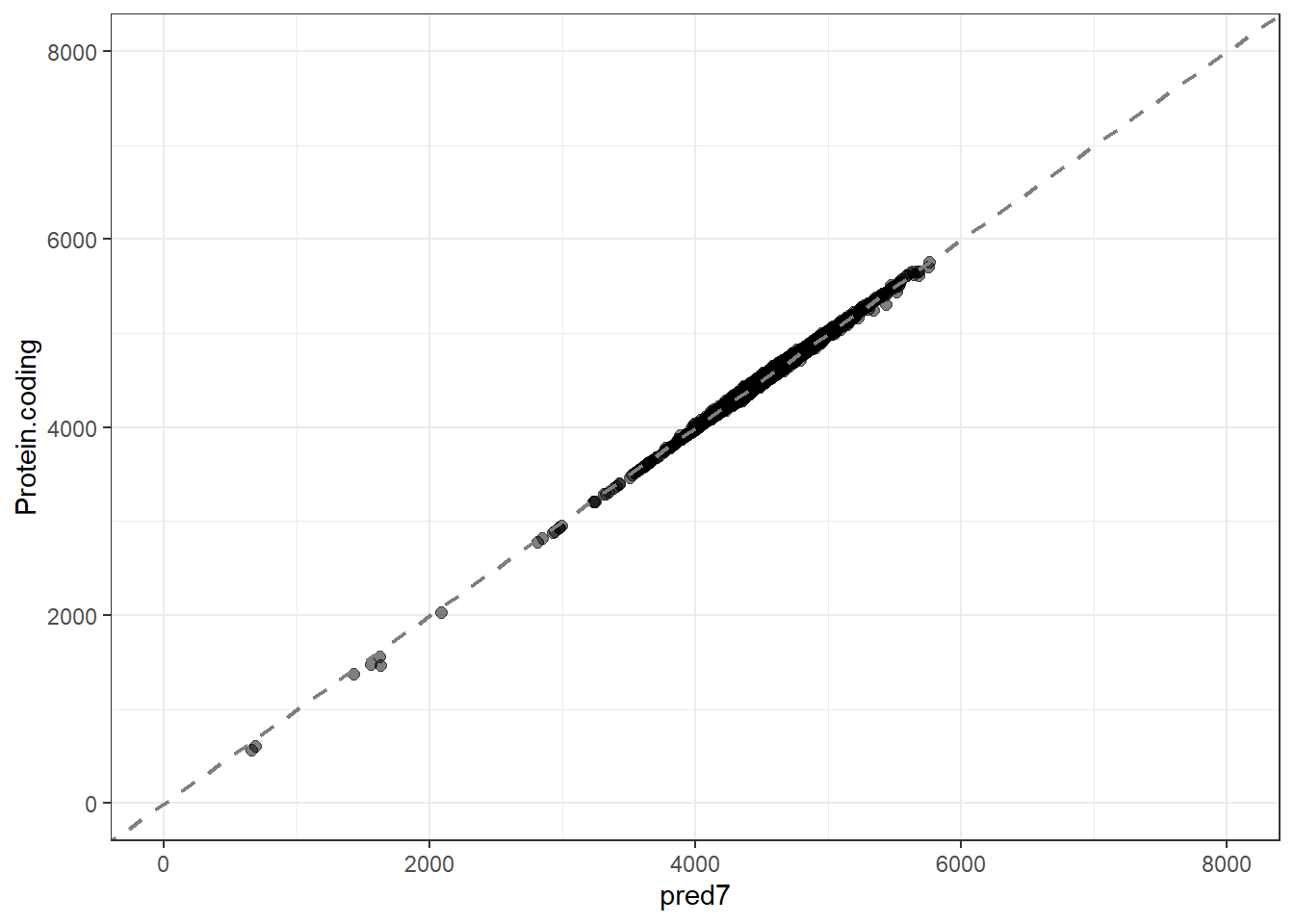
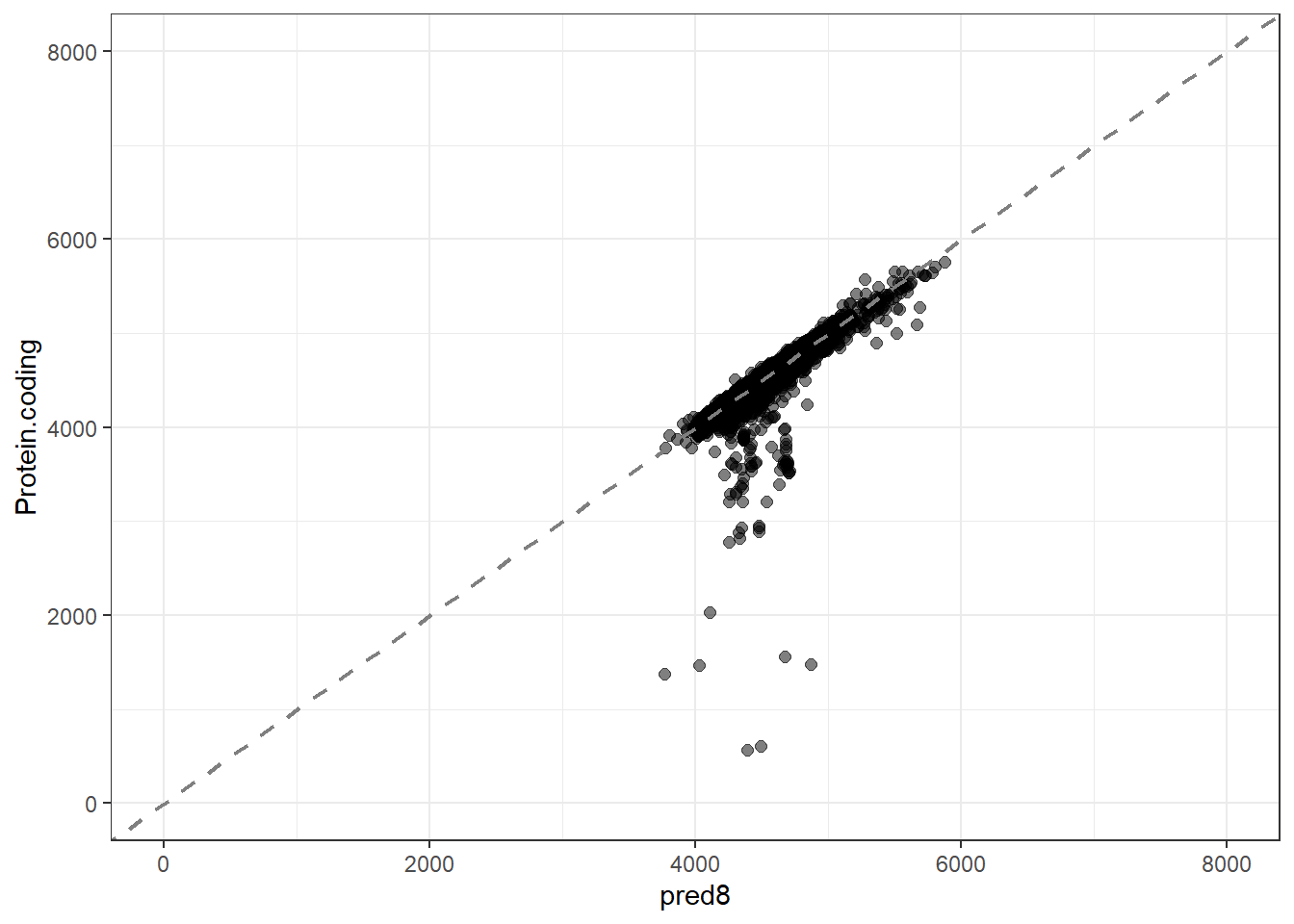
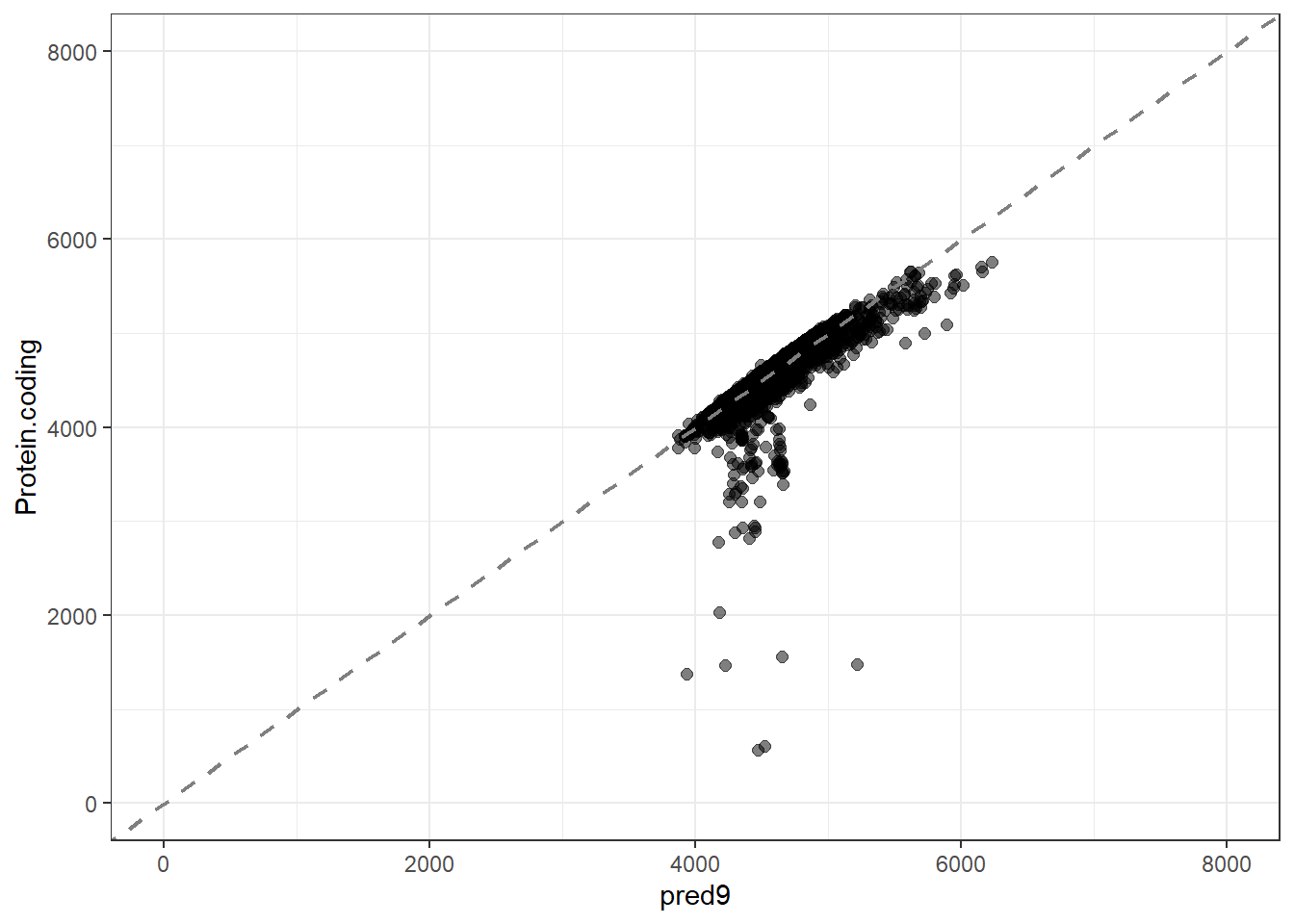
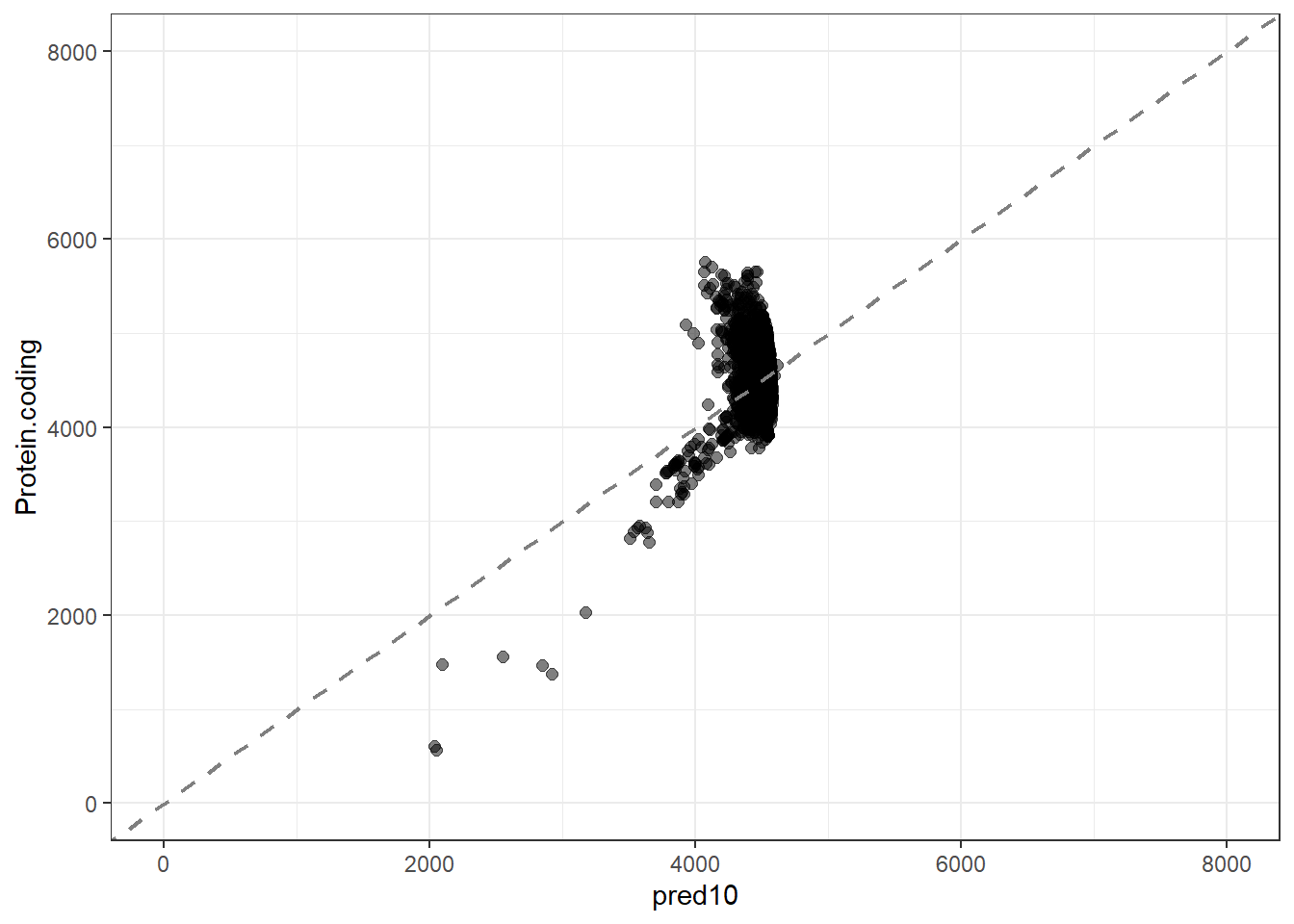
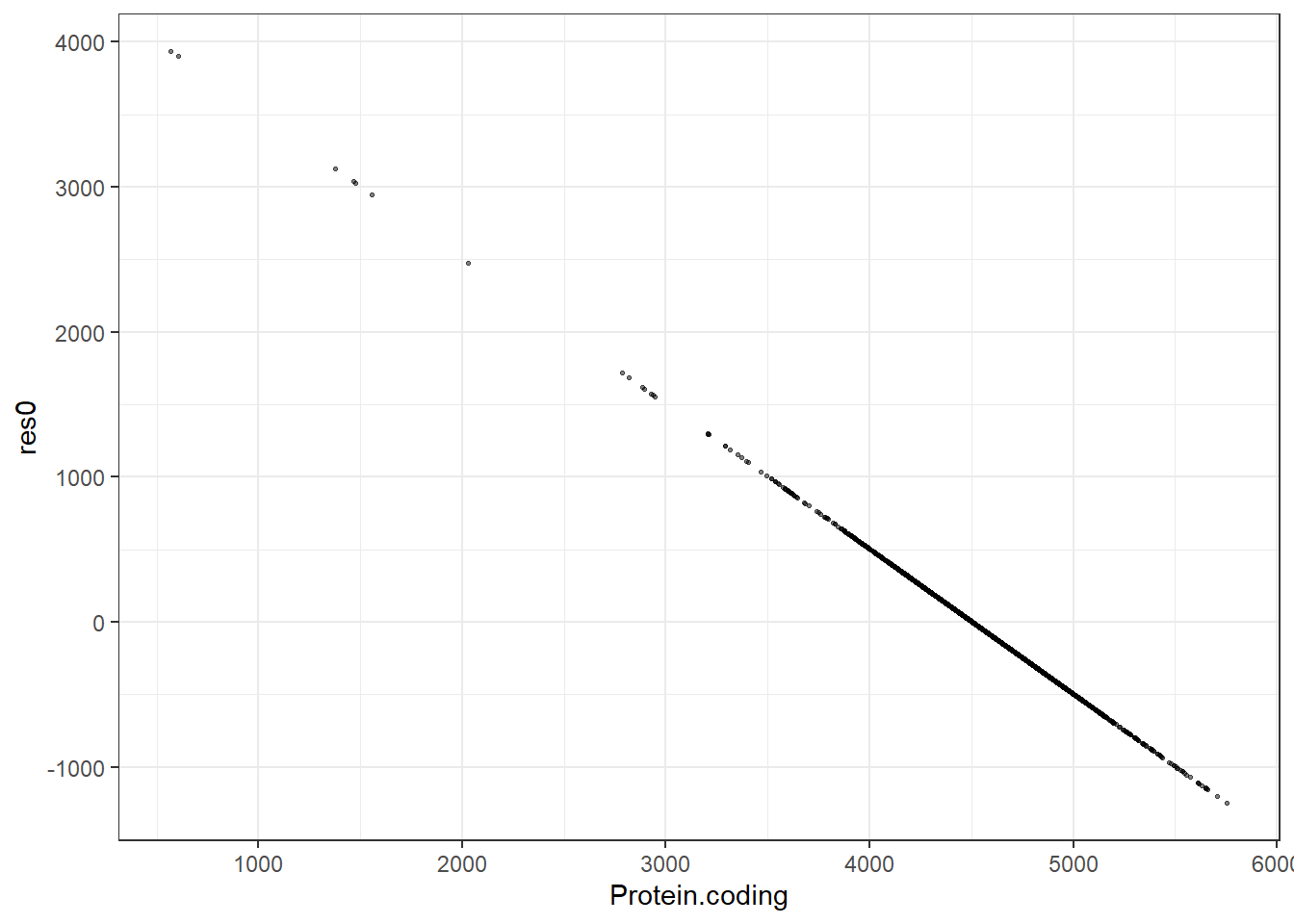
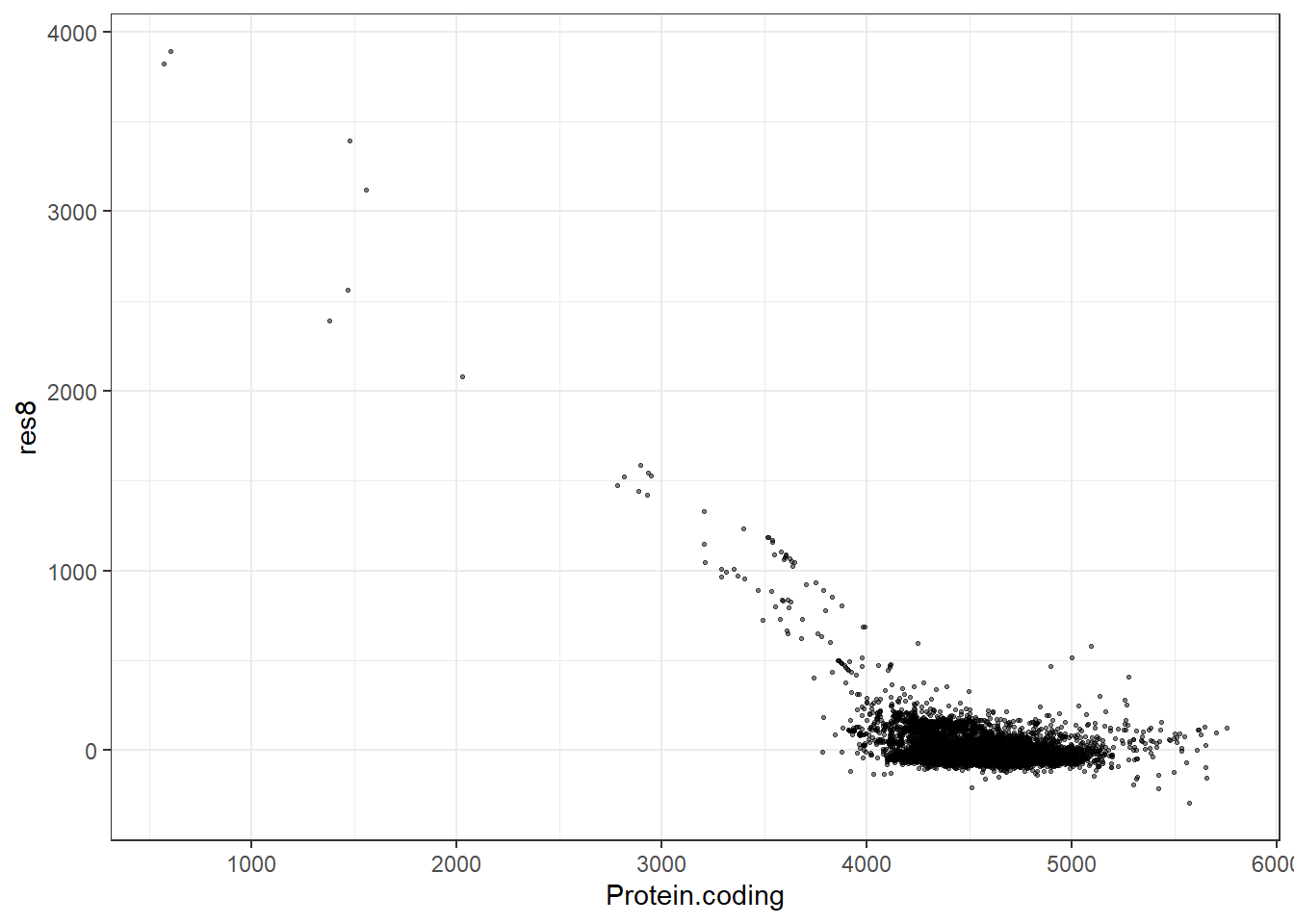
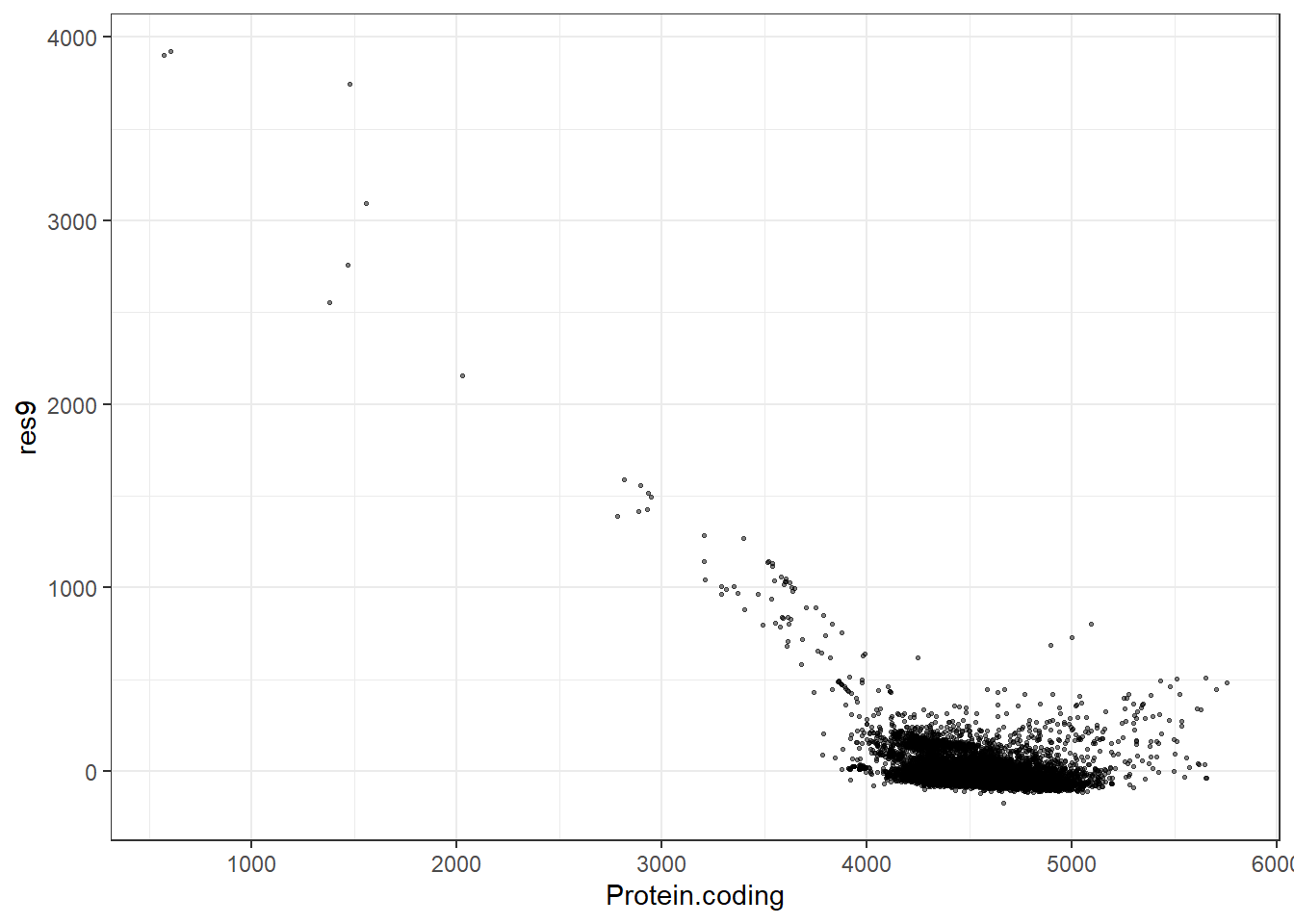
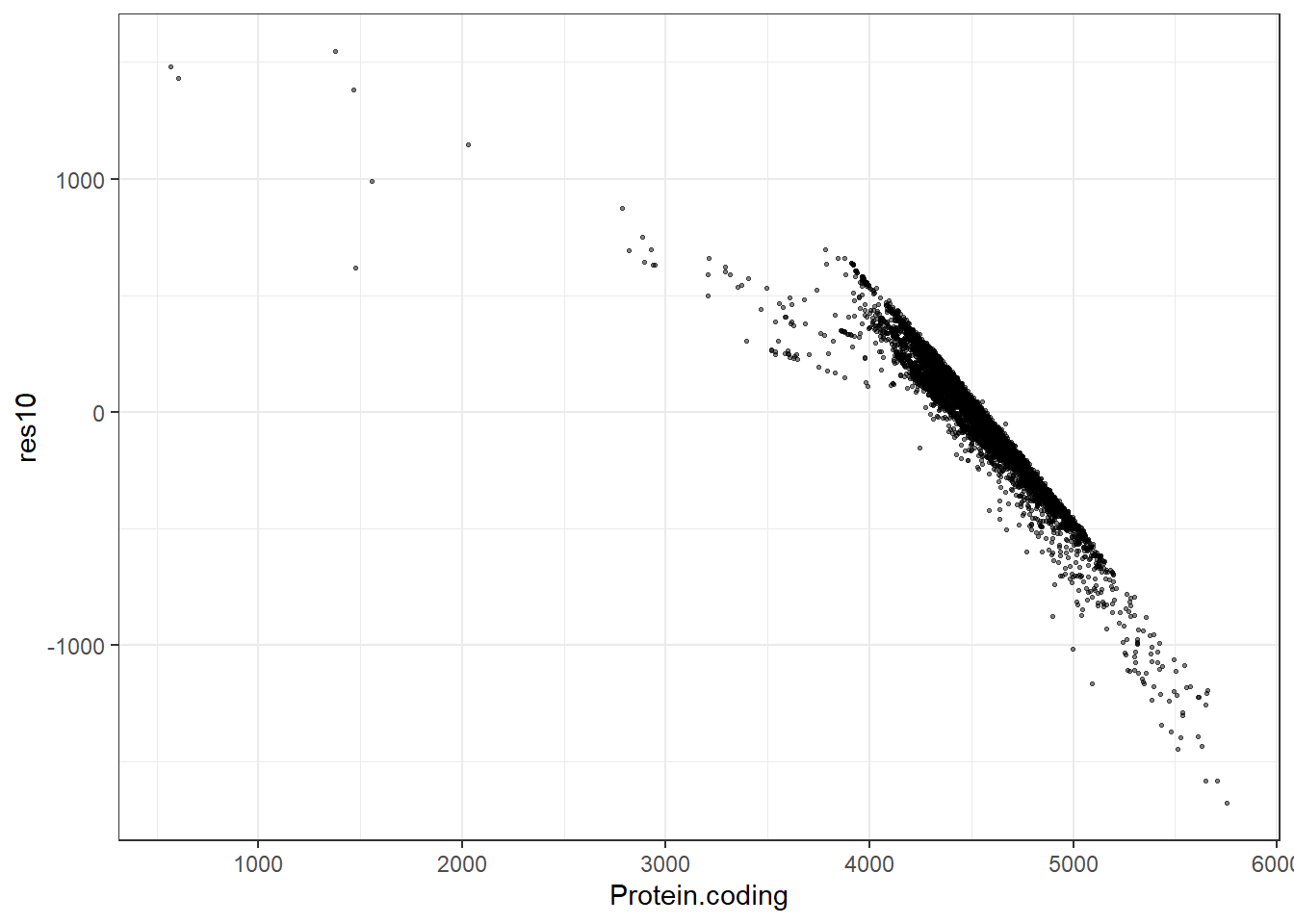
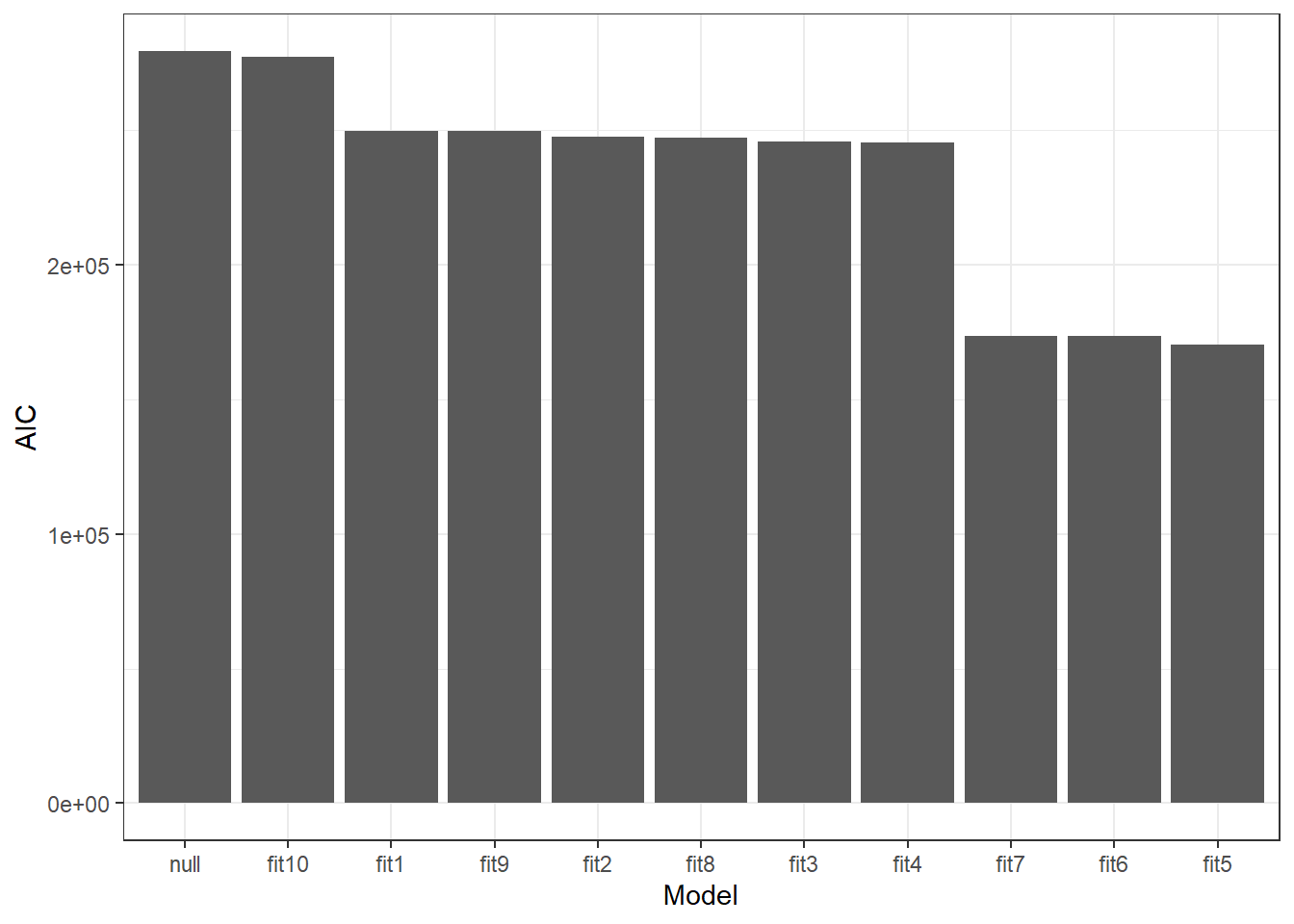
#Predidiction and differences for each fit
genomes1 <- genomes1 %>%
mutate(
pred0 = predict(lm_null, new_data = genomes1, type = "numeric")$.pred,
pred1 = predict(lm_fit1, new_data = genomes1, type = "numeric")$.pred,
pred2 = predict(lm_fit2, new_data = genomes1, type = "numeric")$.pred,
pred3 = predict(lm_fit3, new_data = genomes1, type = "numeric")$.pred,
pred4 = predict(lm_fit4, new_data = genomes1, type = "numeric")$.pred,
pred5 = predict(lm_fit5, new_data = genomes1, type = "numeric")$.pred,
pred6 = predict(lm_fit6, new_data = genomes1, type = "numeric")$.pred,
pred7 = predict(lm_fit7, new_data = genomes1, type = "numeric")$.pred,
pred8 = predict(lm_fit8, new_data = genomes1, type = "numeric")$.pred,
pred9 = predict(lm_fit9, new_data = genomes1, type = "numeric")$.pred,
pred10 = predict(lm_fit10, new_data = genomes1, type = "numeric")$.pred
) %>%
mutate(
res0 = pred0 - Protein.coding,
res1 = pred1 - Protein.coding,
res2 = pred2 - Protein.coding,
res3 = pred3 - Protein.coding,
res4 = pred4 - Protein.coding,
res5 = pred5 - Protein.coding,
res6 = pred6 - Protein.coding,
res7 = pred7 - Protein.coding,
res8 = pred8 - Protein.coding,
res9 = pred9 - Protein.coding,
res10 = pred10 - Protein.coding
)