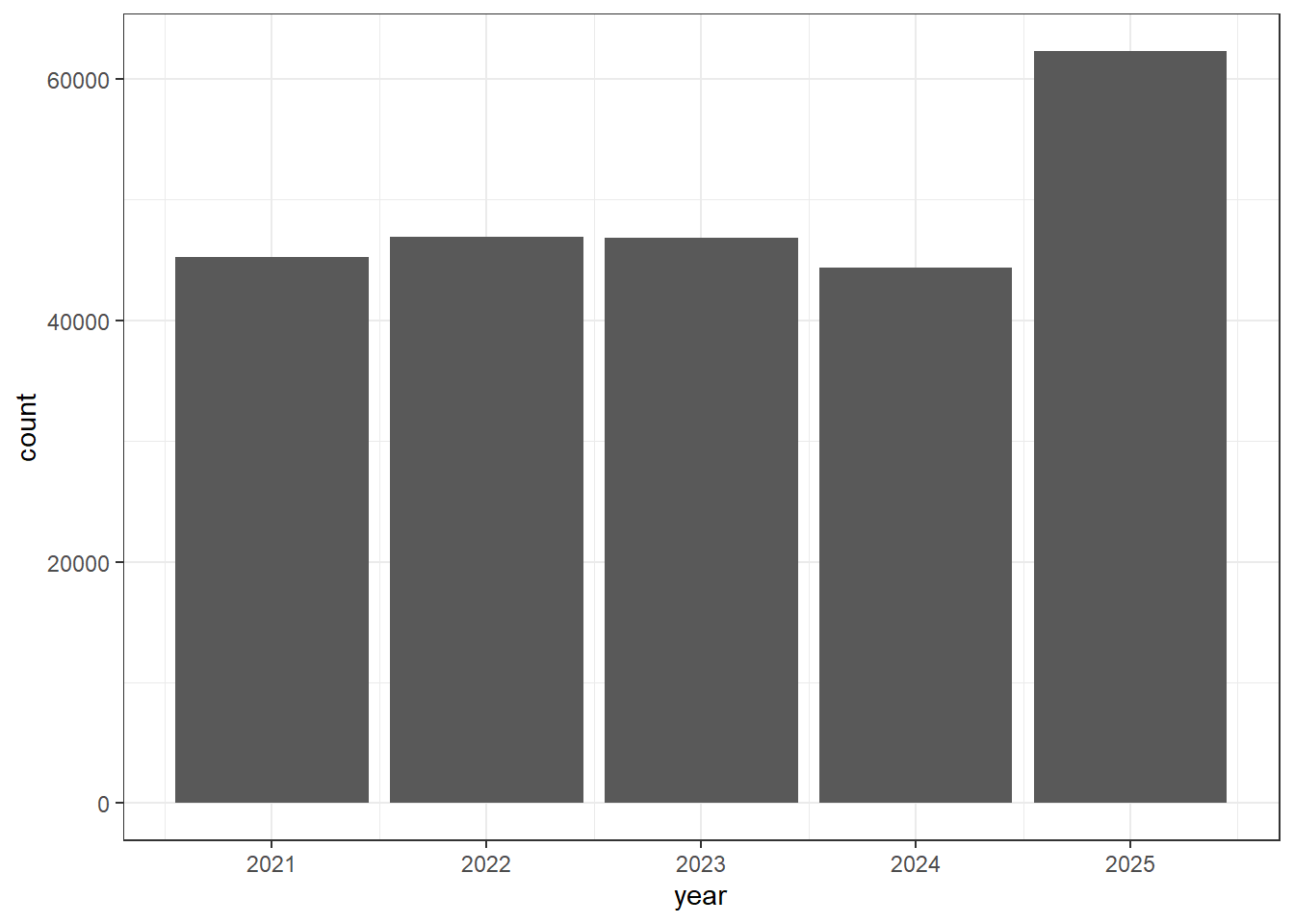
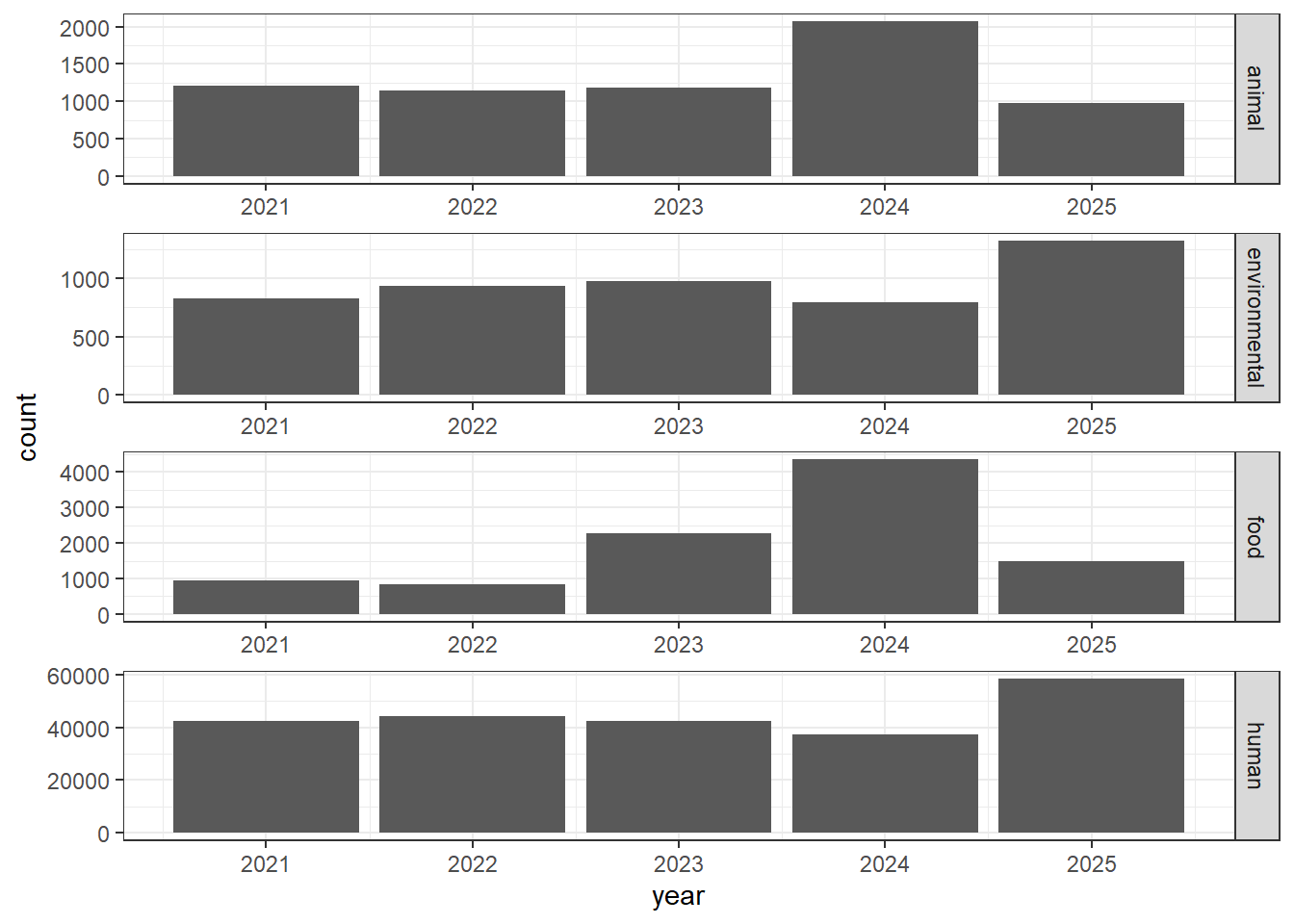
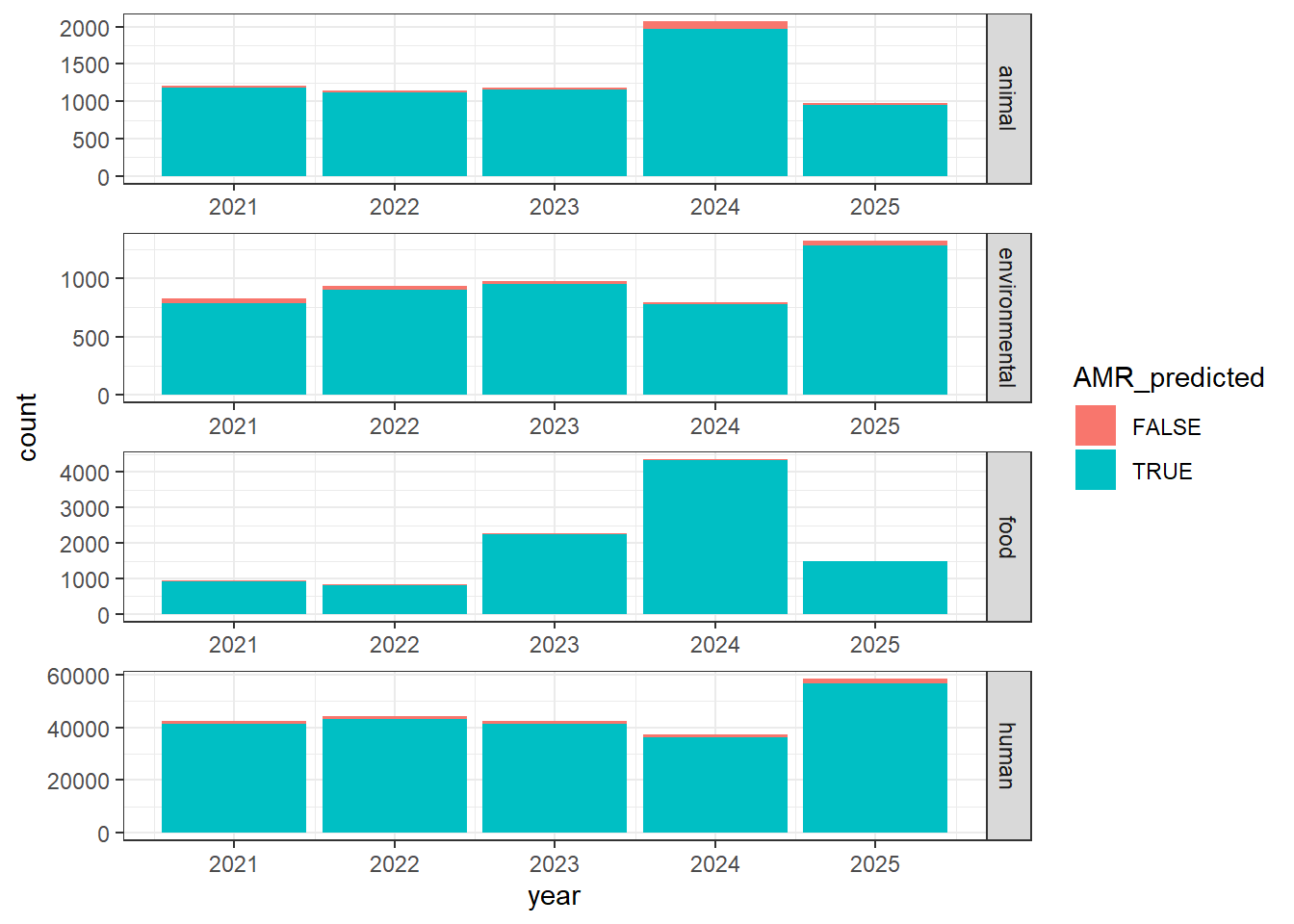
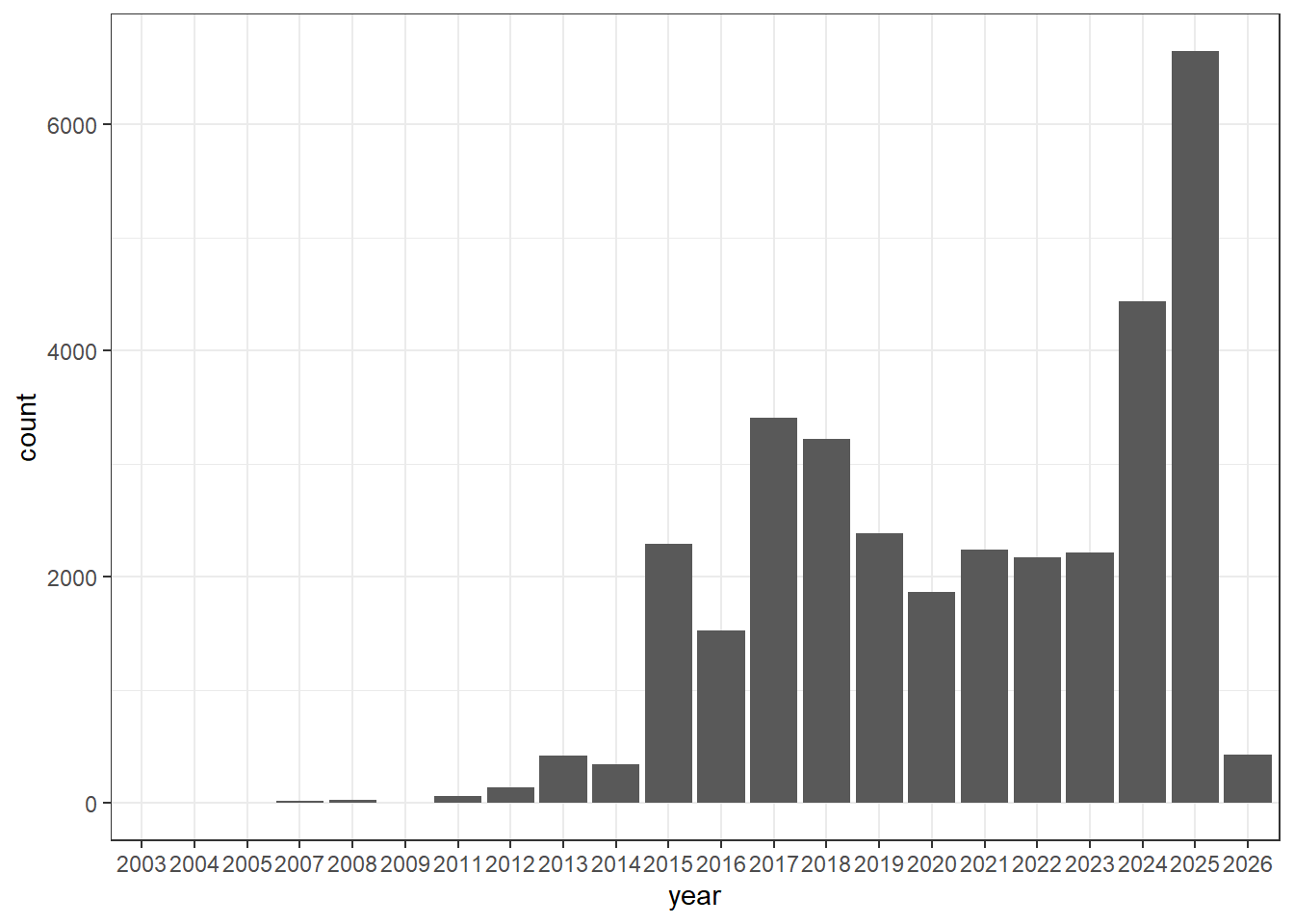
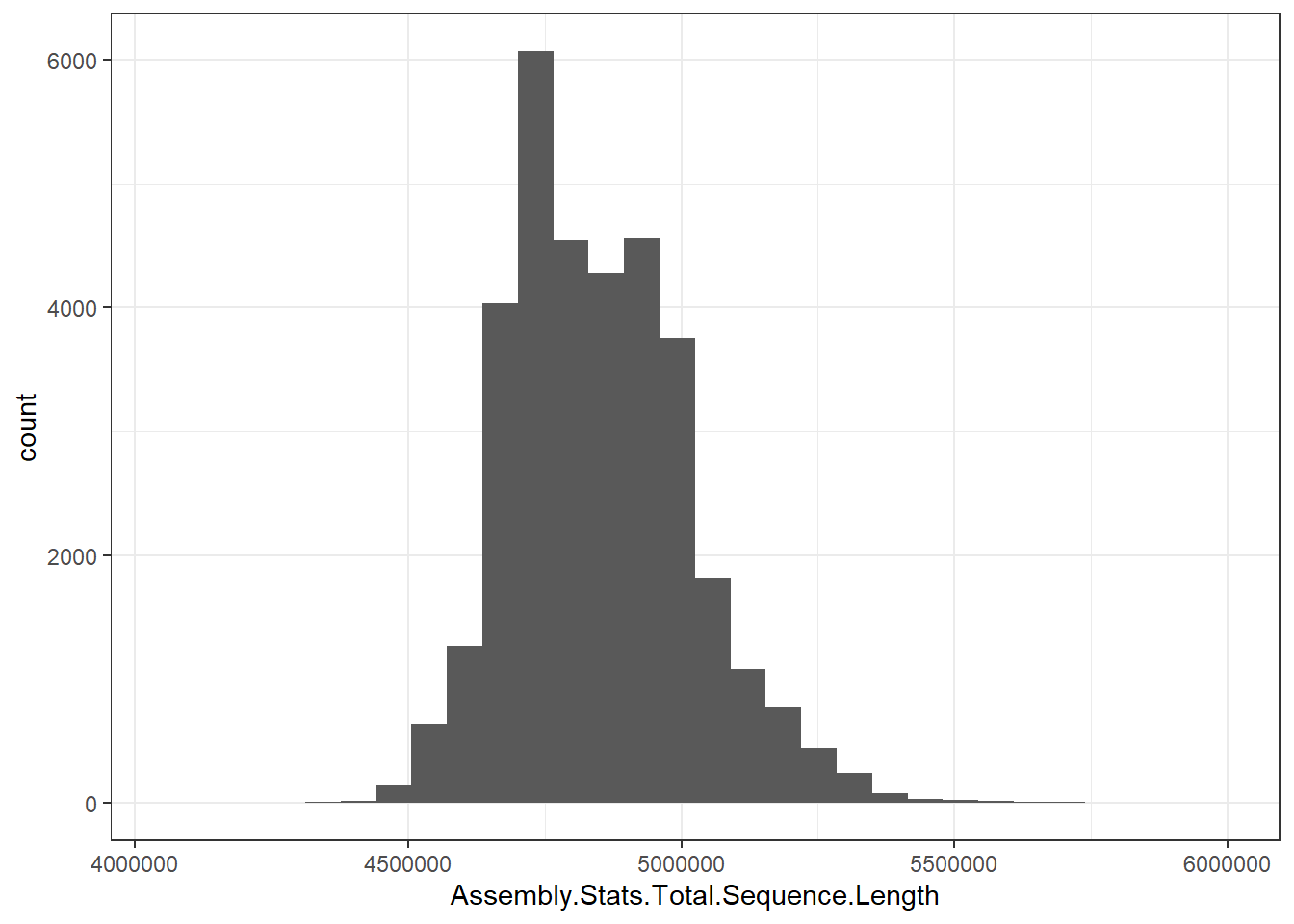
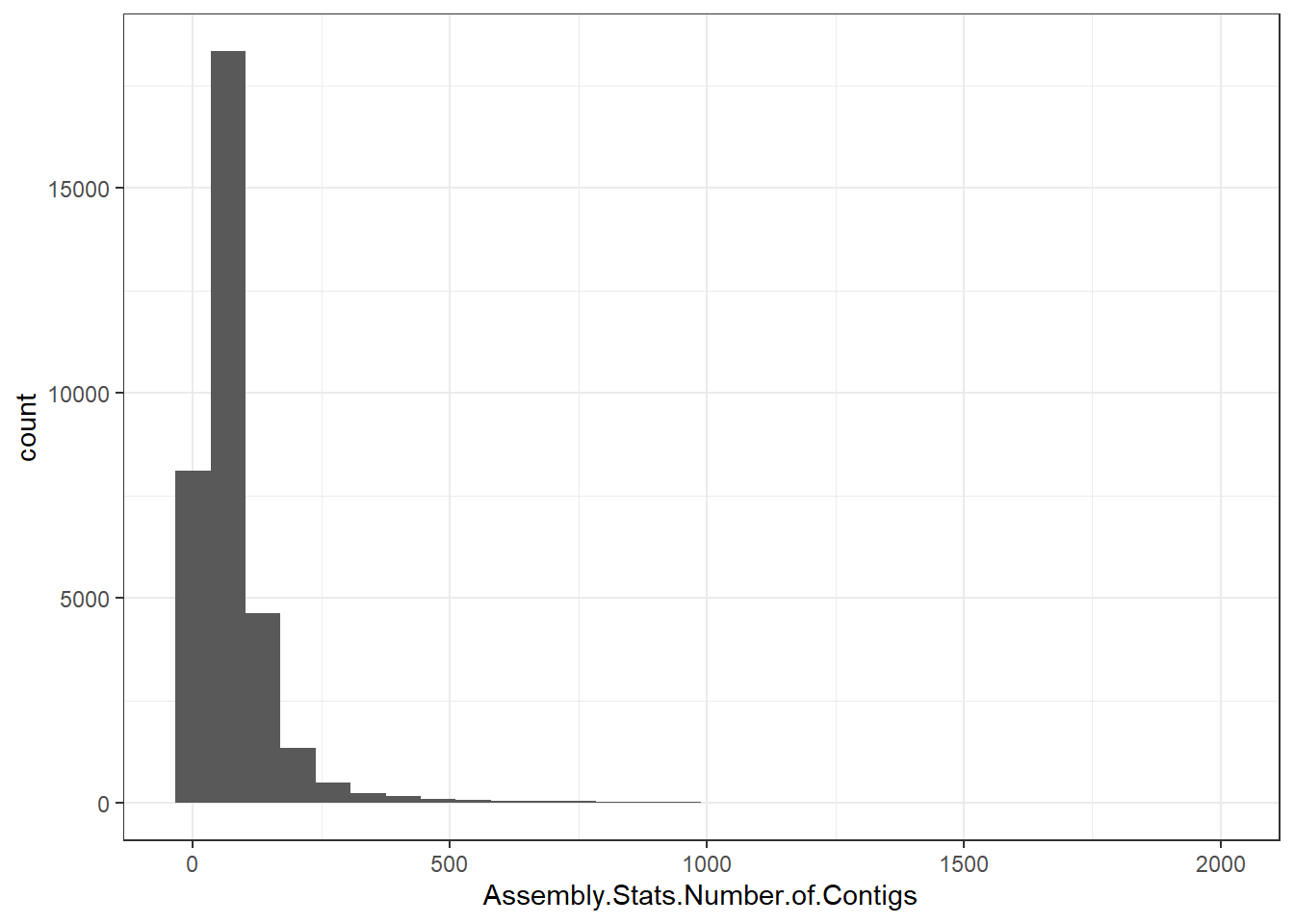
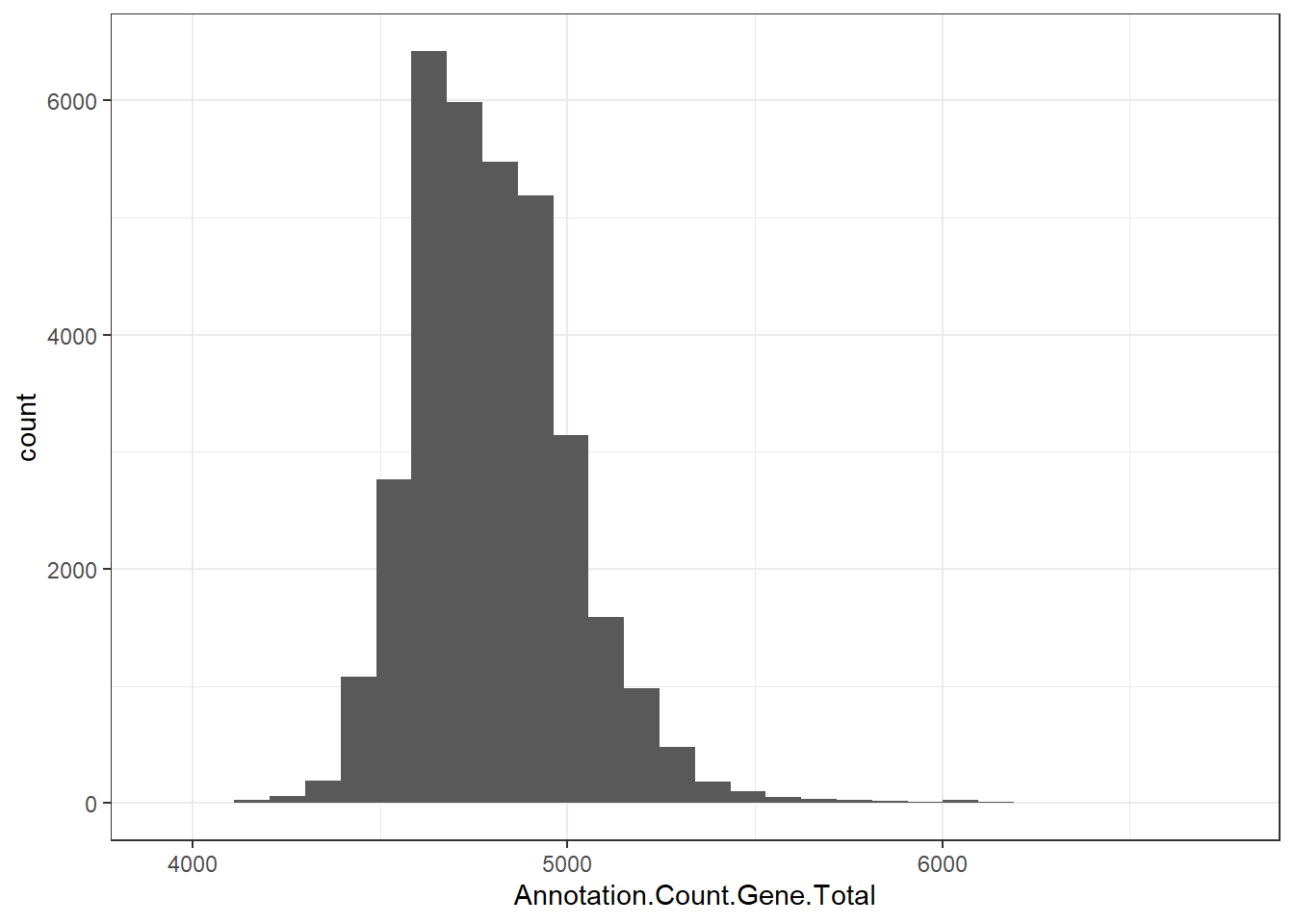
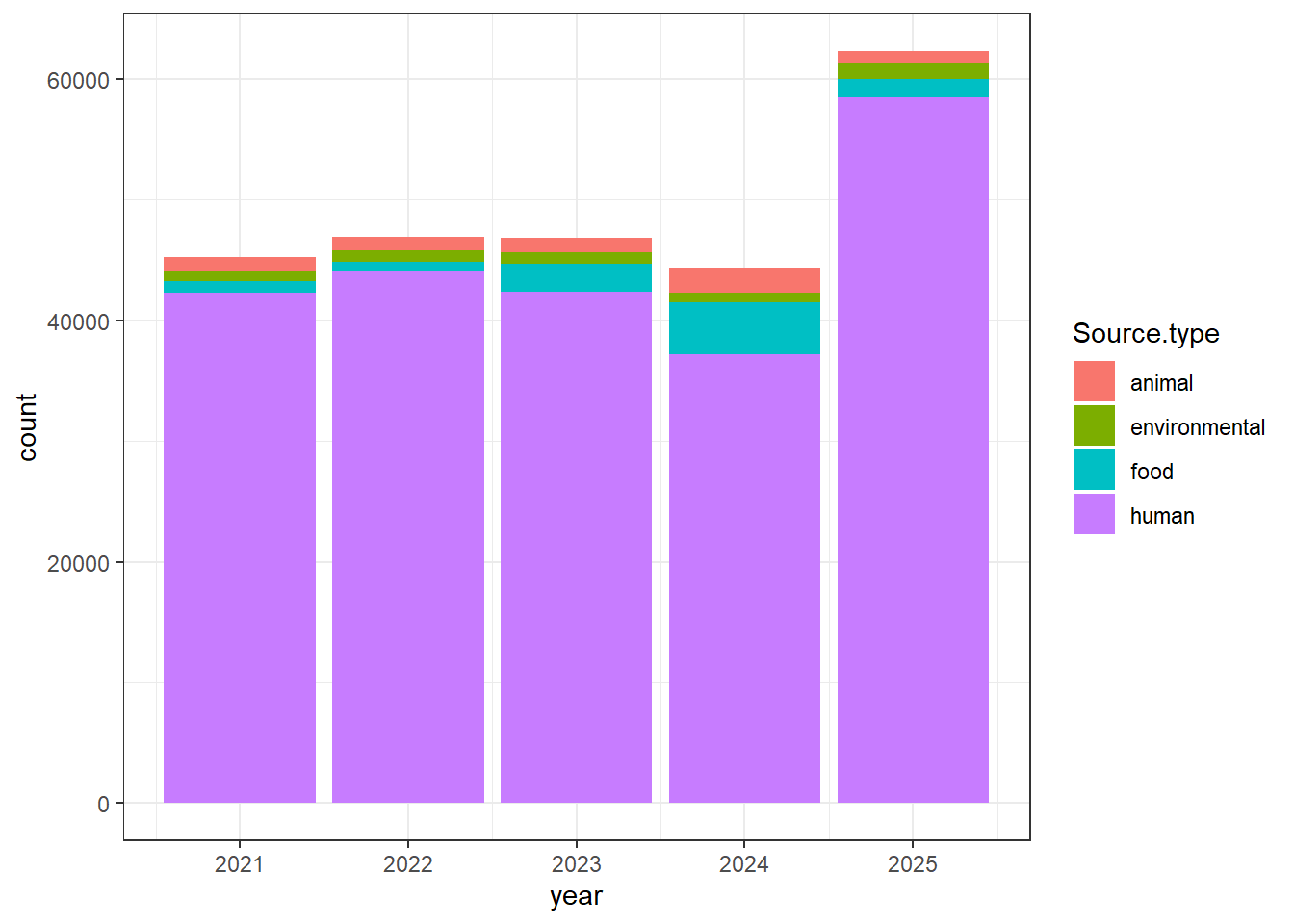#Isolates obtained from _NCBI Pathogen Detection_ database: Salmonella
isolates_raw <- read_tsv(here("data/raw-data", "isolates.tsv"))
#Changing format from tsv to csv
write_csv(isolates_raw, here("data/raw-data/", "isolates0.csv"))
#Making a new object based on new csv
isolates0 <- read.csv(here("data/raw-data/", "isolates0.csv"))
#Genomes dataset from NCBI: _Bacterial genomes_
genomes_raw <- read_tsv(here("data/raw-data/", "genomes_datasets.tsv"))
#Changing format from tsv to csv
write_csv(genomes_raw, here("data/raw-data/", "genomes0.csv"))
#Making a new object based on new csv
genomes0 <- read.csv(here("data/raw-data/", "genomes0.csv"))
#Removing non useful columns from isolates0
isolates0$WGS.accession = NULL
isolates0$WGS.prefix = NULL
isolates0$PFGE.secondary.enzyme.pattern = NULL
isolates0$PFGE.primary.enzyme.pattern = NULL
isolates0$Platform = NULL
isolates0$Host.disease = NULL
isolates0$Outbreak = NULL
isolates0$SRA.release.date = NULL
isolates0$SRA.Center = NULL
isolates0$Run = NULL
isolates0$Library.layout = NULL
isolates0$Lat.Lon = NULL
isolates0$AST.phenotypes = NULL
isolates0$AMR.genotypes.core = NULL
isolates0$Host = NULL
isolates0$Min.same = NULL
isolates0$Min.diff = NULL
#Removing non-useful columns from genomes0
genomes0$Assembly.Paired.Assembly.Accession = NULL
genomes0$Organism.Infraspecific.Names.Breed = NULL
genomes0$Organism.Infraspecific.Names.Strain = NULL
genomes0$Organism.Infraspecific.Names.Cultivar = NULL
genomes0$Organism.Infraspecific.Names.Ecotype = NULL
genomes0$Organism.Infraspecific.Names.Isolate = NULL
genomes0$Organism.Infraspecific.Names.Sex = NULL
genomes0$WGS.project.accession = NULL
genomes0$Annotation.BUSCO.Complete = NULL
genomes0$Annotation.BUSCO.Single.Copy = NULL
genomes0$Annotation.BUSCO.Duplicated = NULL
genomes0$Annotation.BUSCO.Fragmented = NULL
genomes0$Annotation.BUSCO.Missing = NULL
genomes0$Annotation.BUSCO.Lineage = NULL
genomes0$Type.Material.Display.Text = NULL
genomes0$CheckM.marker.set = NULL
genomes0$CheckM.completeness = NULL
genomes0$CheckM.contamination = NULL
genomes0$slm_filter = NULL
genomes0$release_year = NULL
#Creating BioSample column
genomes0$BioSample = genomes0$Assembly.BioSample.Accession
#Both dataframes are still too large >100 MB they will be streamlined even more by keeping only selected columns
#Selected columns for isolates dataframe
isolates1 <- data.frame("Create.date" = isolates0$Create.date,
"Location" = isolates0$Location,
"Isolation.source" = isolates0$Isolation.source,
"Source.type" = isolates0$Source.type,
"BioSample" = isolates0$BioSample,
"AMR.genotypes" = isolates0$AMR.genotypes,
"Computed.types" = isolates0$Computed.types
)
#Identifying country names where isolates came from to NCBI
isolates1$country_name <- country_name(x= isolates1$Location, to="ISO3")
#Subsetting only for isolates obtained in the USA in the last 5 complete years
isolates1 <- isolates1[(isolates1$country_name %in% "USA"), ]
isolates1$year <- as.numeric(substr(isolates1$Create.date, 1, 4))
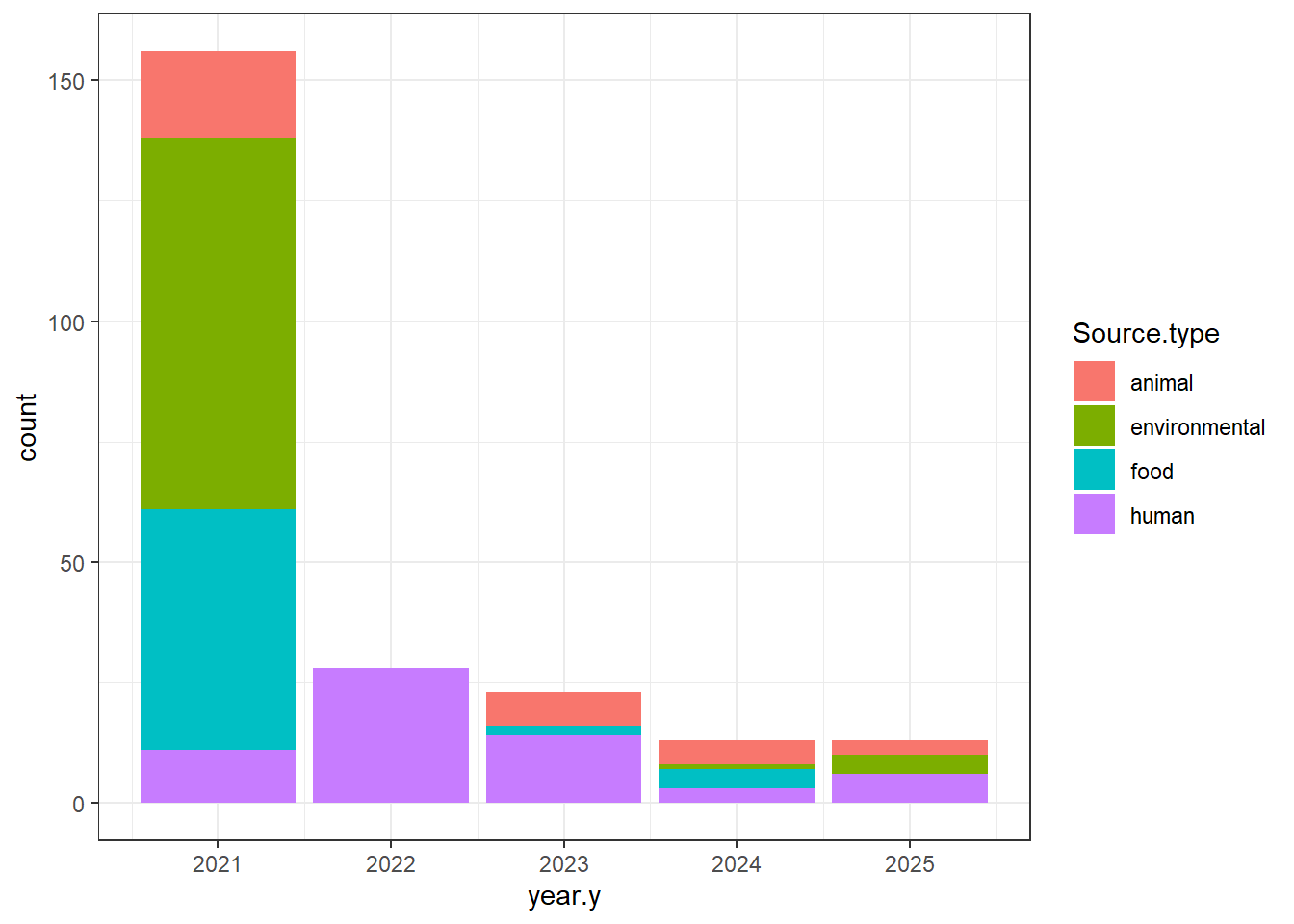
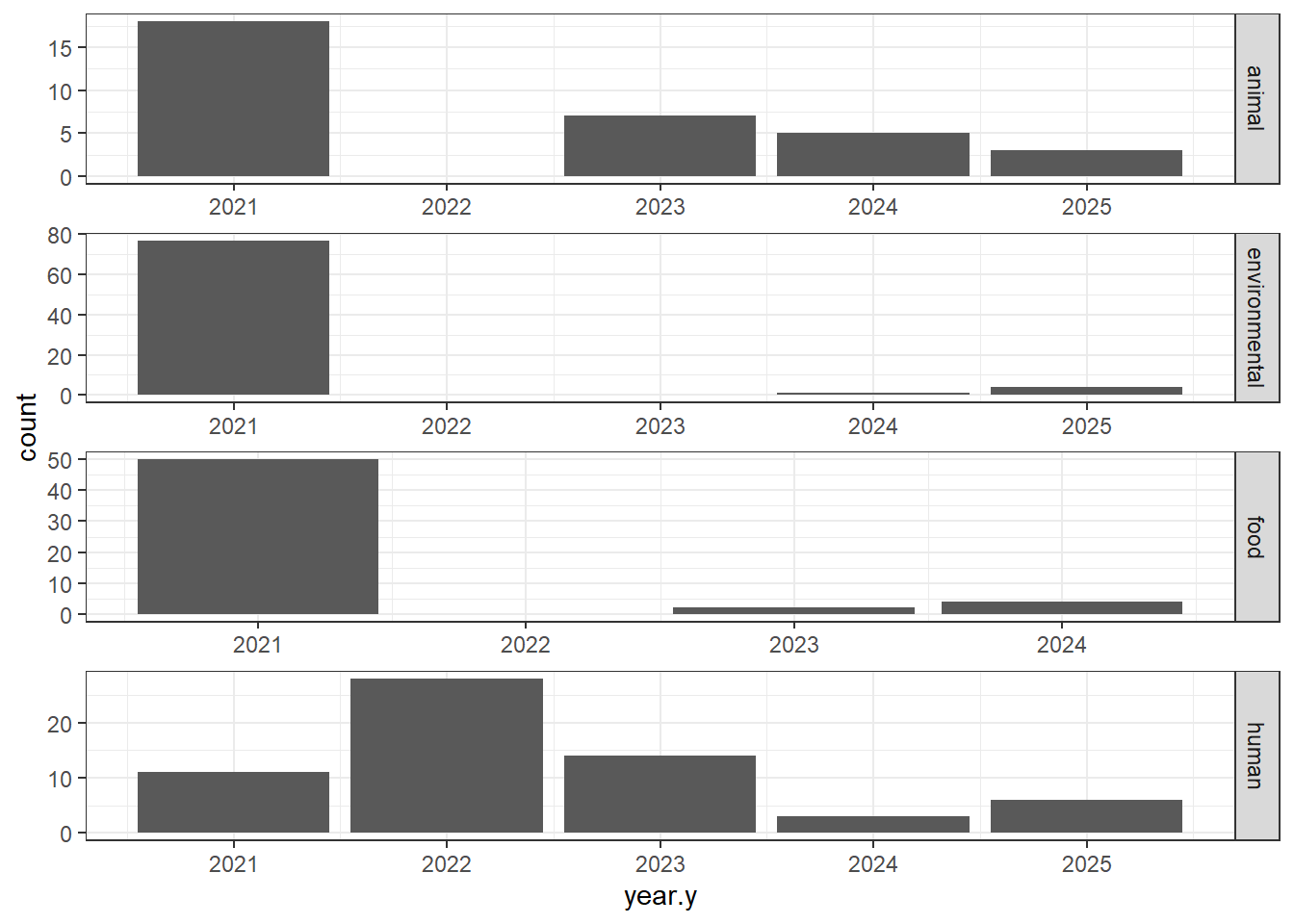
isolates1 <- isolates1[(isolates1$year %in% c(2025, 2024, 2023, 2022, 2021)), ]
#Obtaining only categories of interest
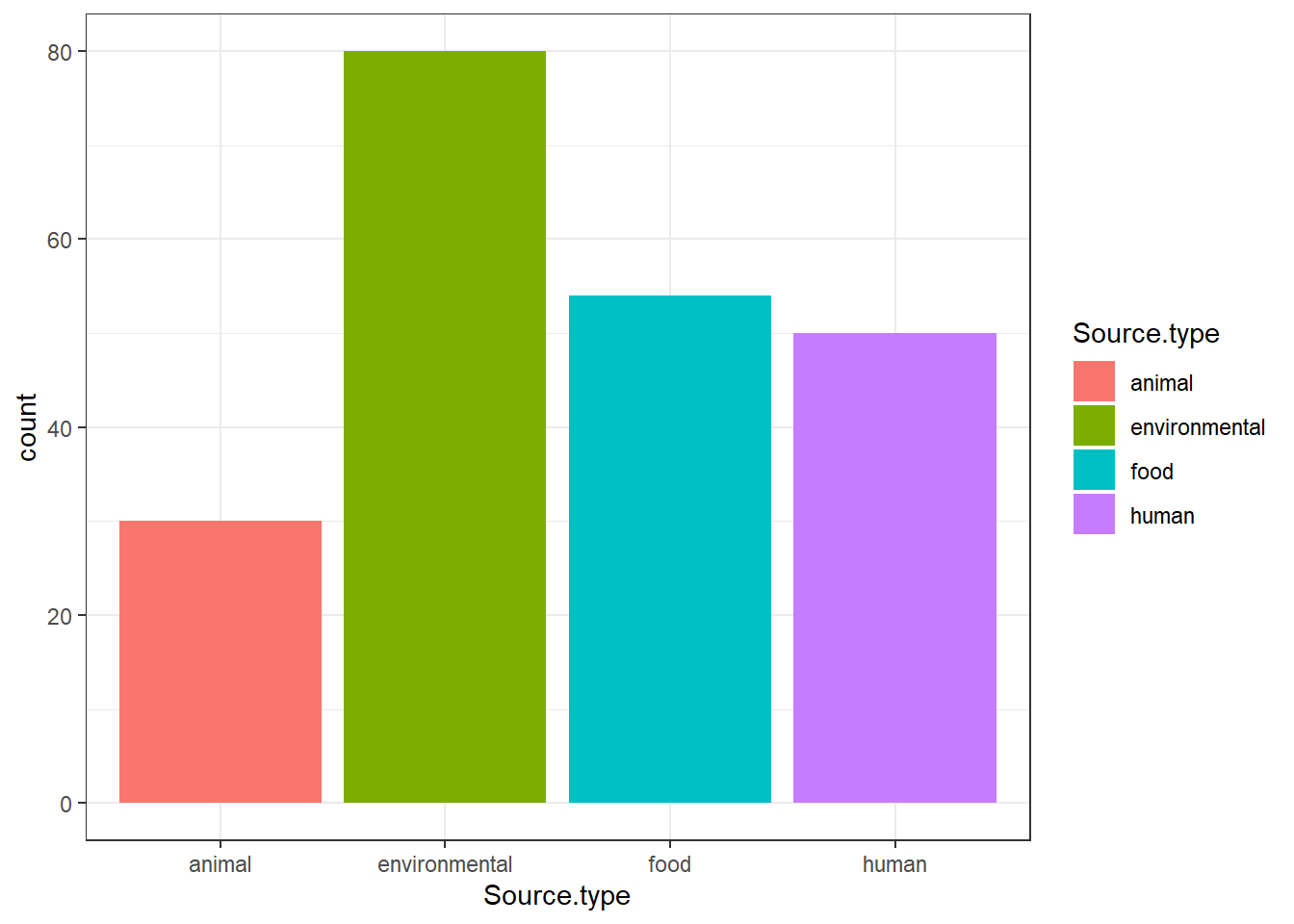
isolates1 <- isolates1[(isolates1$Source.type %in% c("animal", "environmental", "food", "human")), ]
#Saving new df, file under 100 MB
write_csv(isolates1, here("data/processed-data/", "isolates1.csv"))
#Selected colums for genomes dataframe
genomes0$genus <- substr(genomes0$Organism.Name, 1, 10)
genomes1 <- data.frame("Annotation.Release.Date" = genomes0$Annotation.Release.Date,
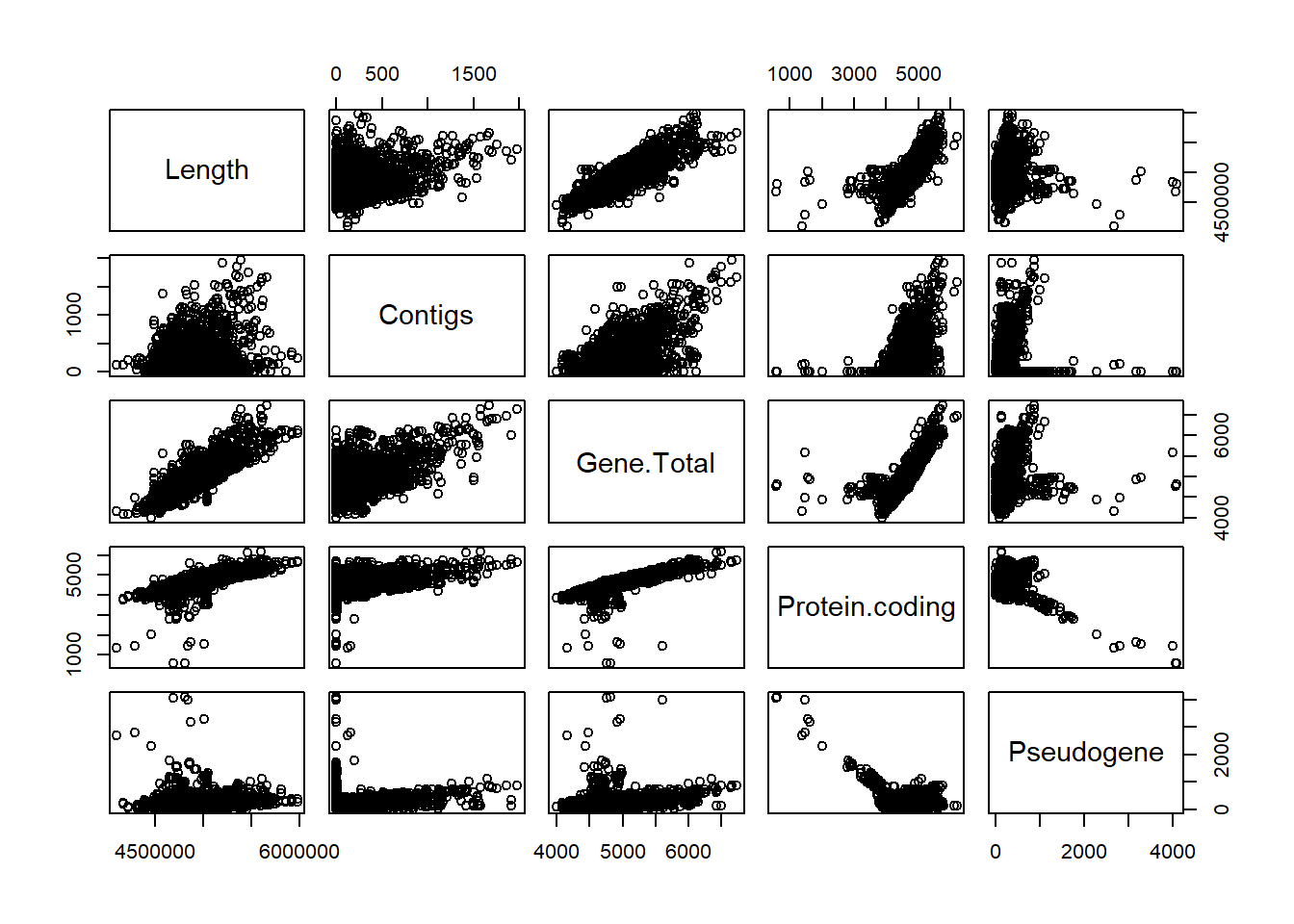
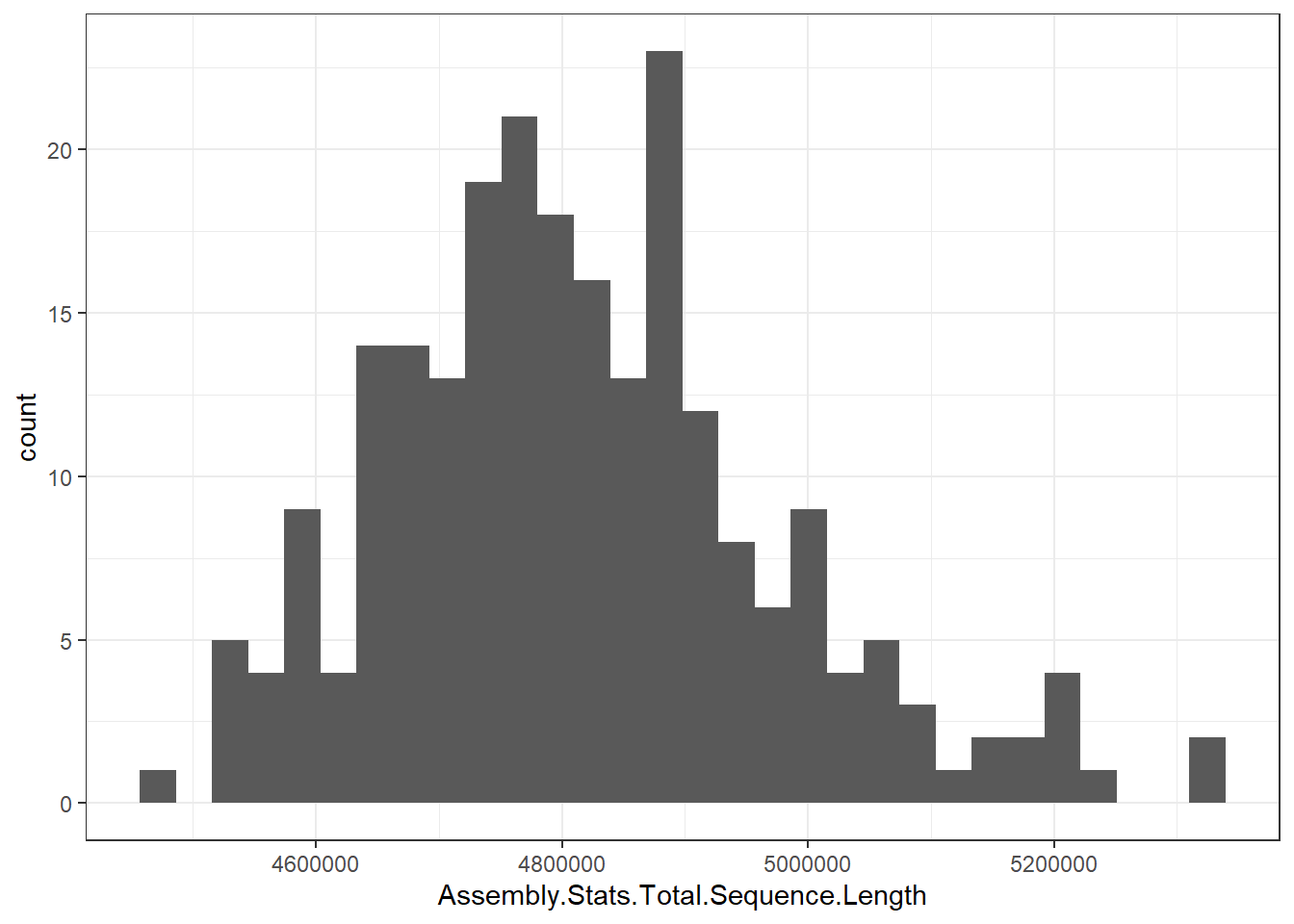
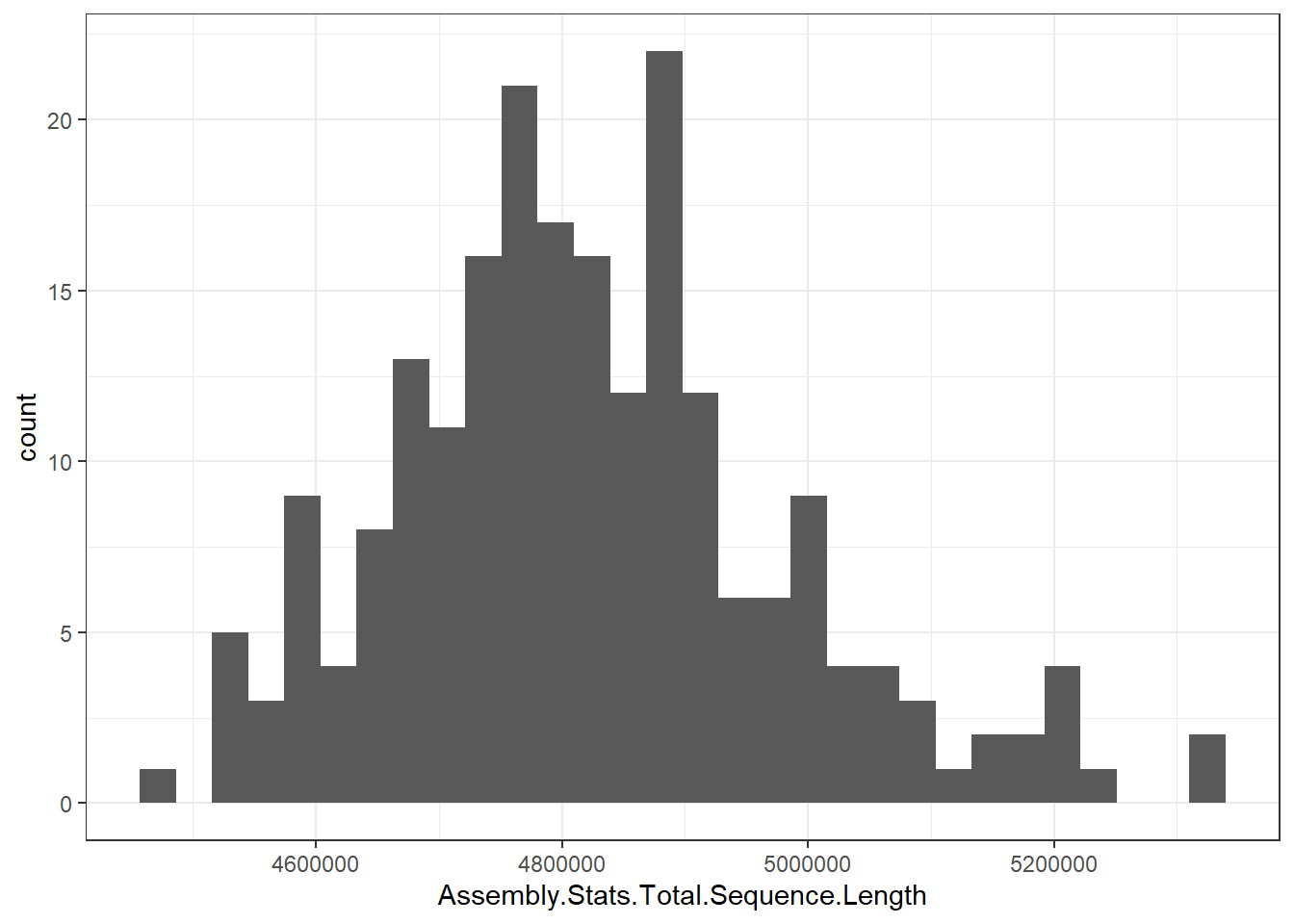
"Assembly.Stats.Total.Sequence.Length" = genomes0$Assembly.Stats.Total.Sequence.Length,
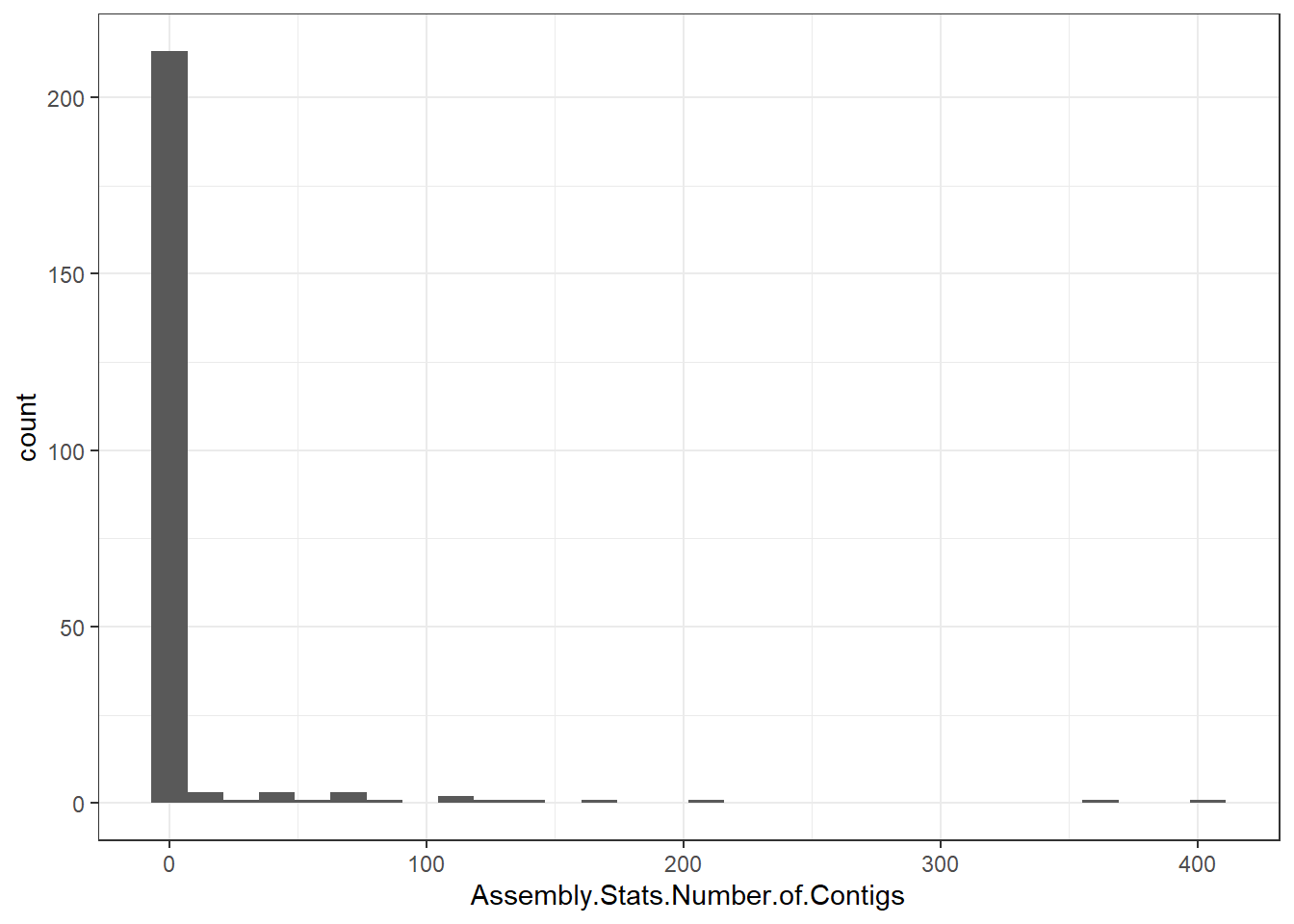
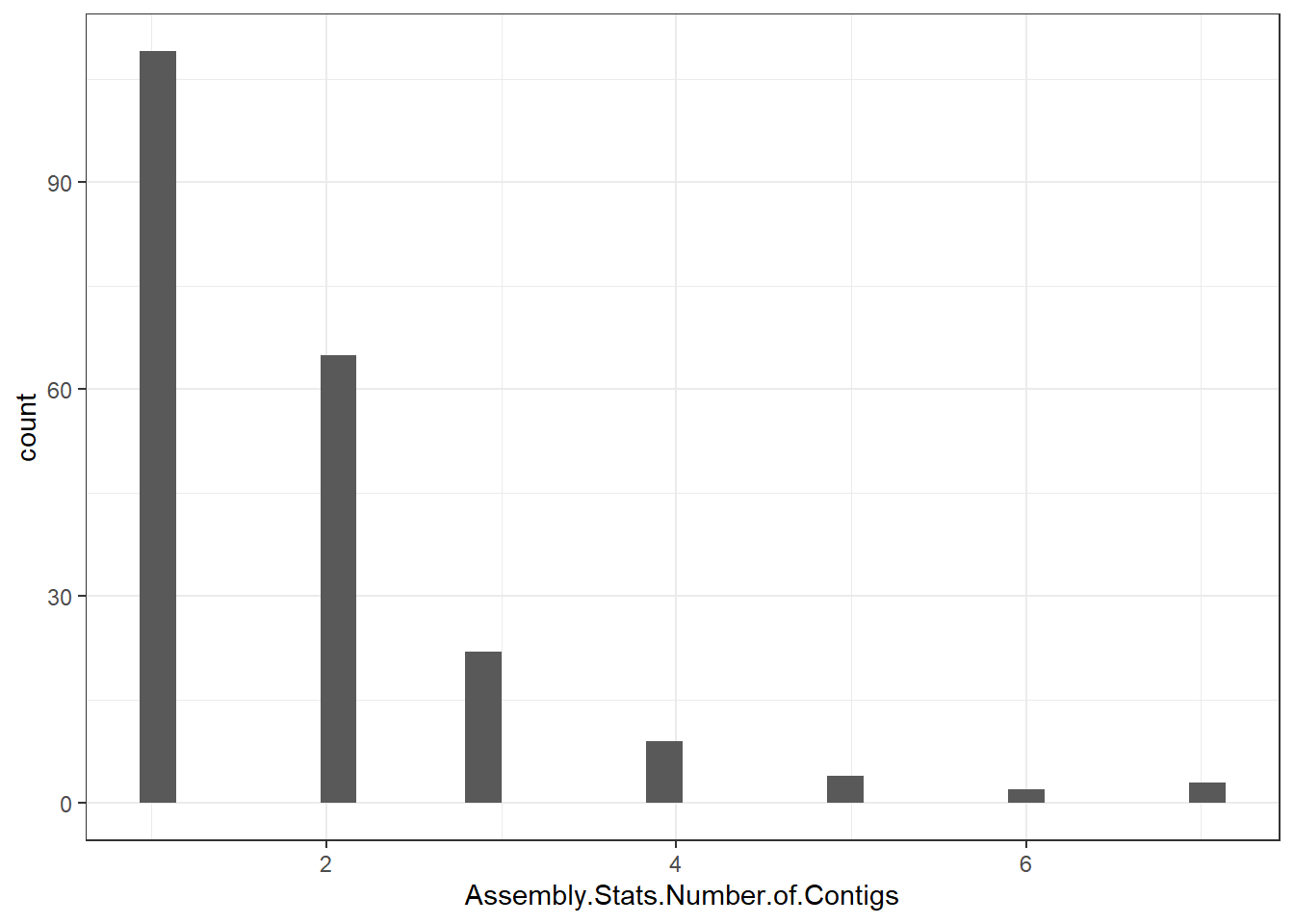
"Assembly.Stats.Number.of.Contigs" = genomes0$Assembly.Stats.Number.of.Contigs,
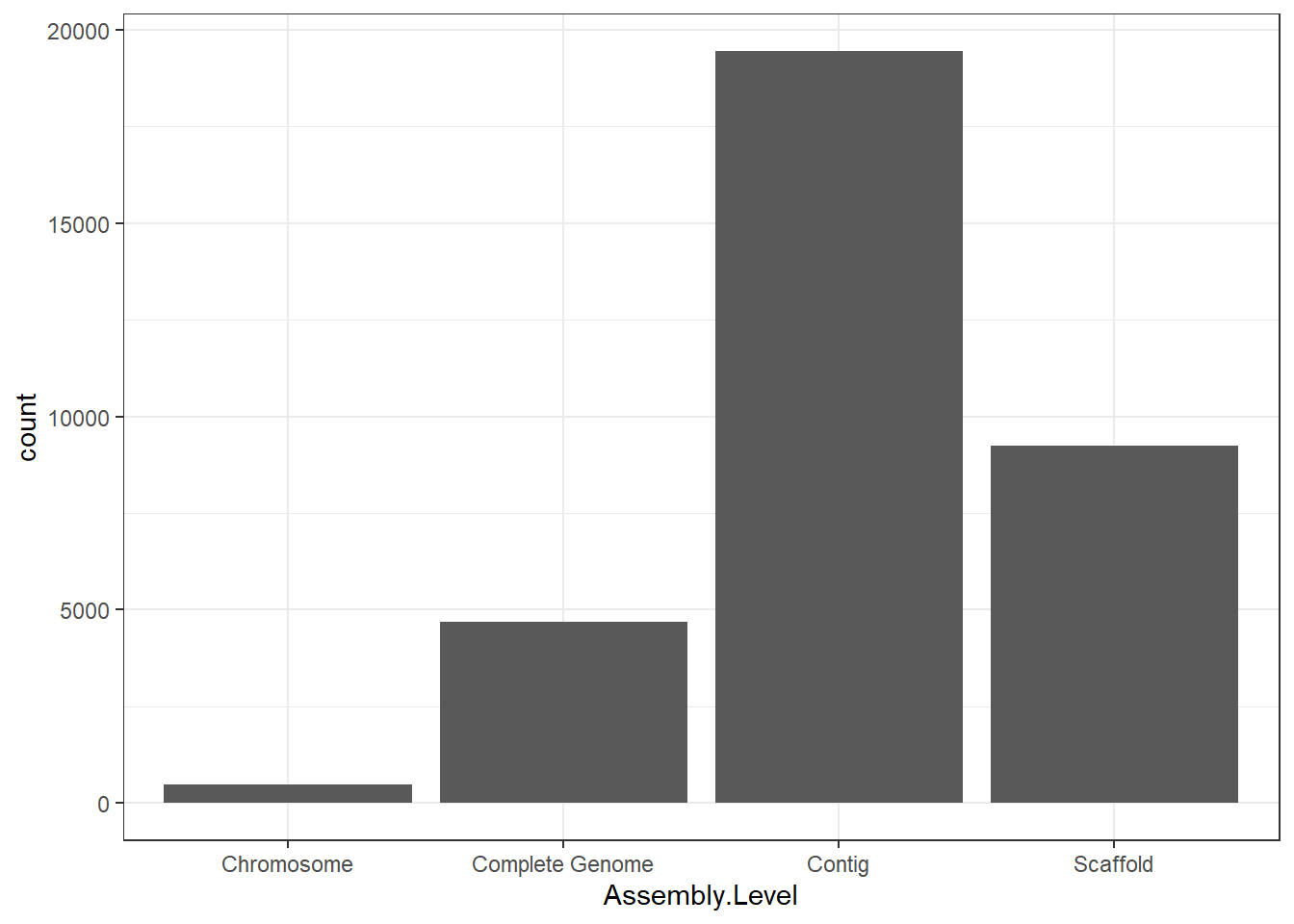
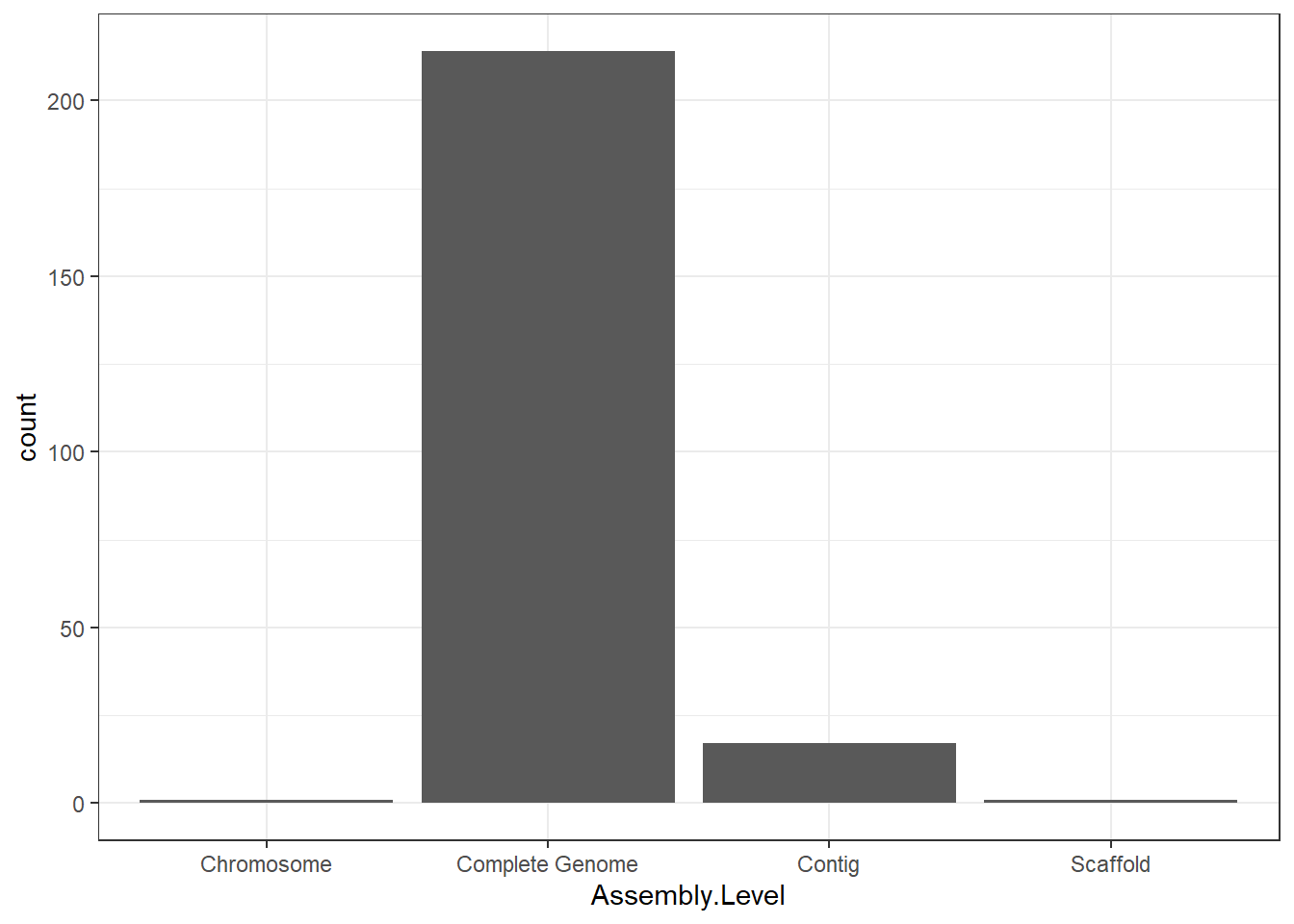
"Assembly.Level" = genomes0$Assembly.Level,
"Assembly.Release.Date" = genomes0$Assembly.Release.Date,
"BioSample" = genomes0$BioSample,
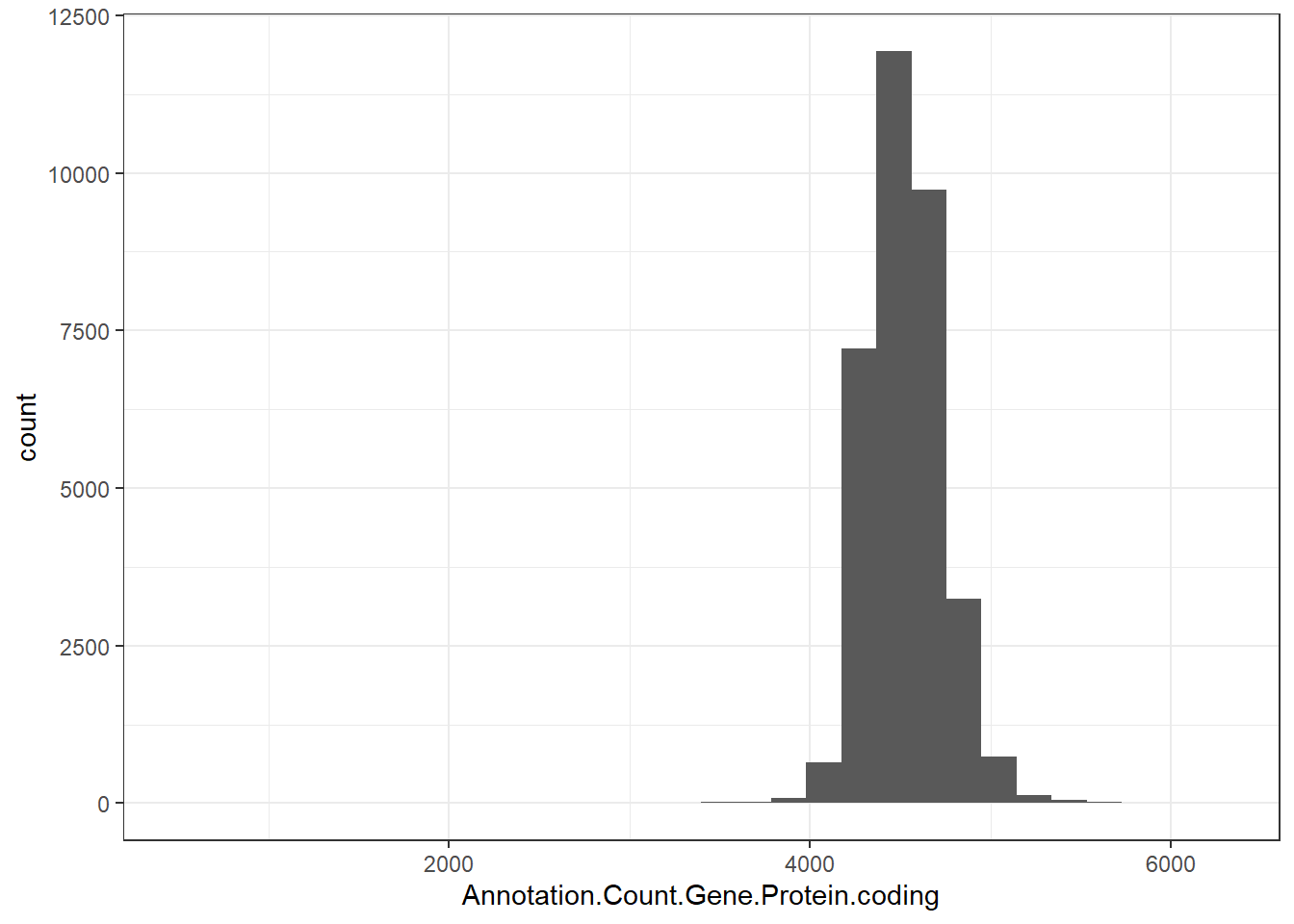
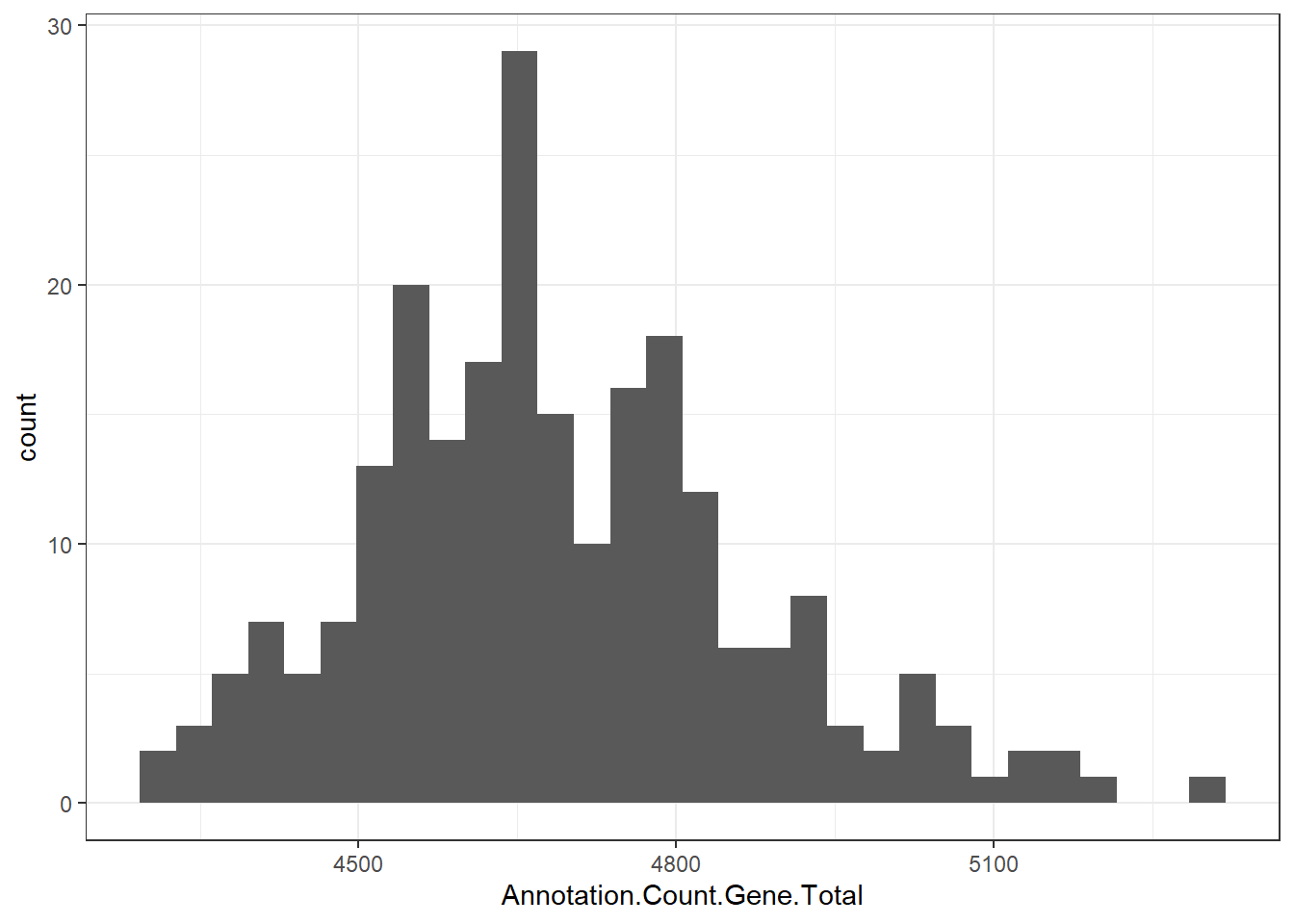
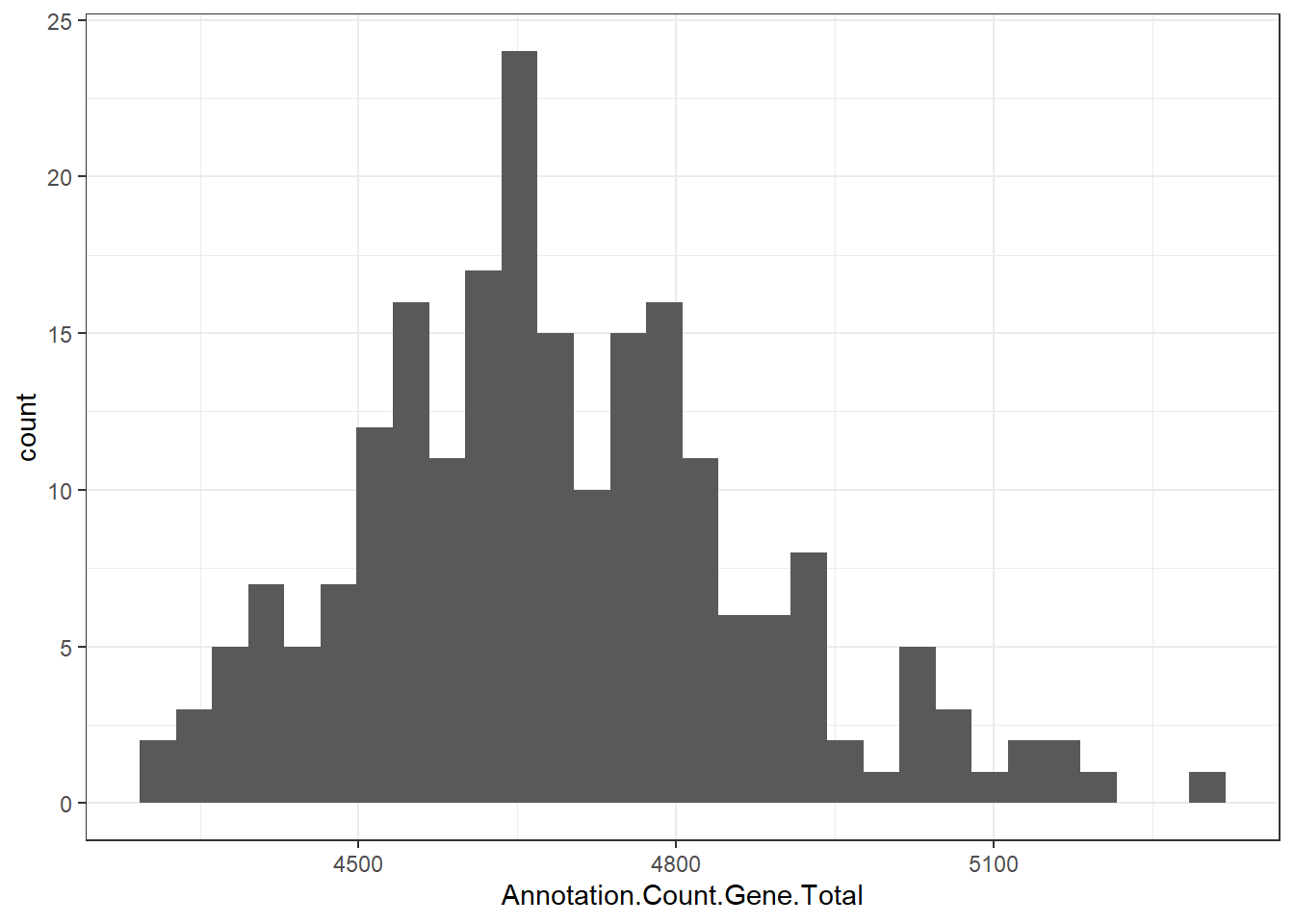
"Annotation.Count.Gene.Total" = genomes0$Annotation.Count.Gene.Total,
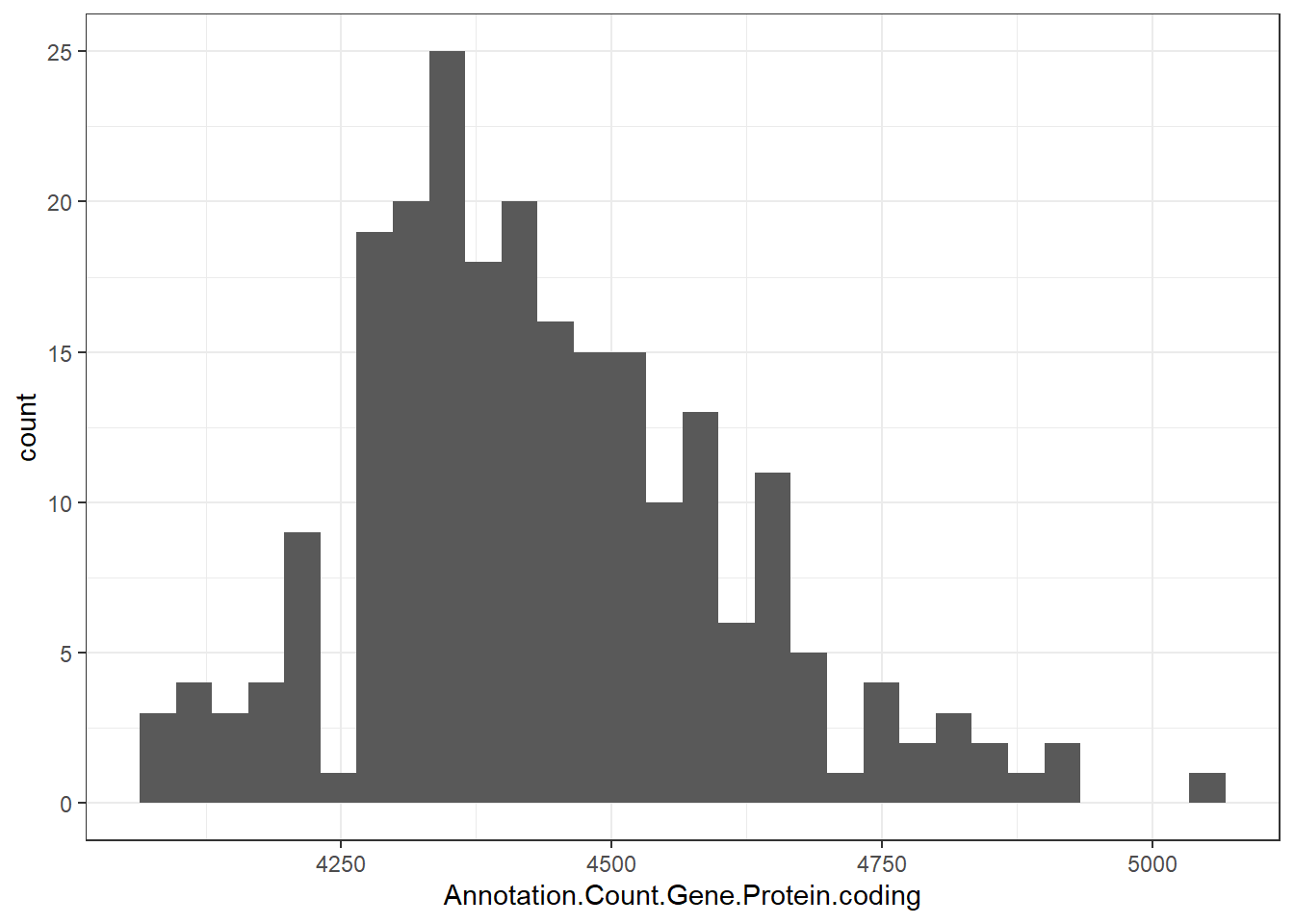
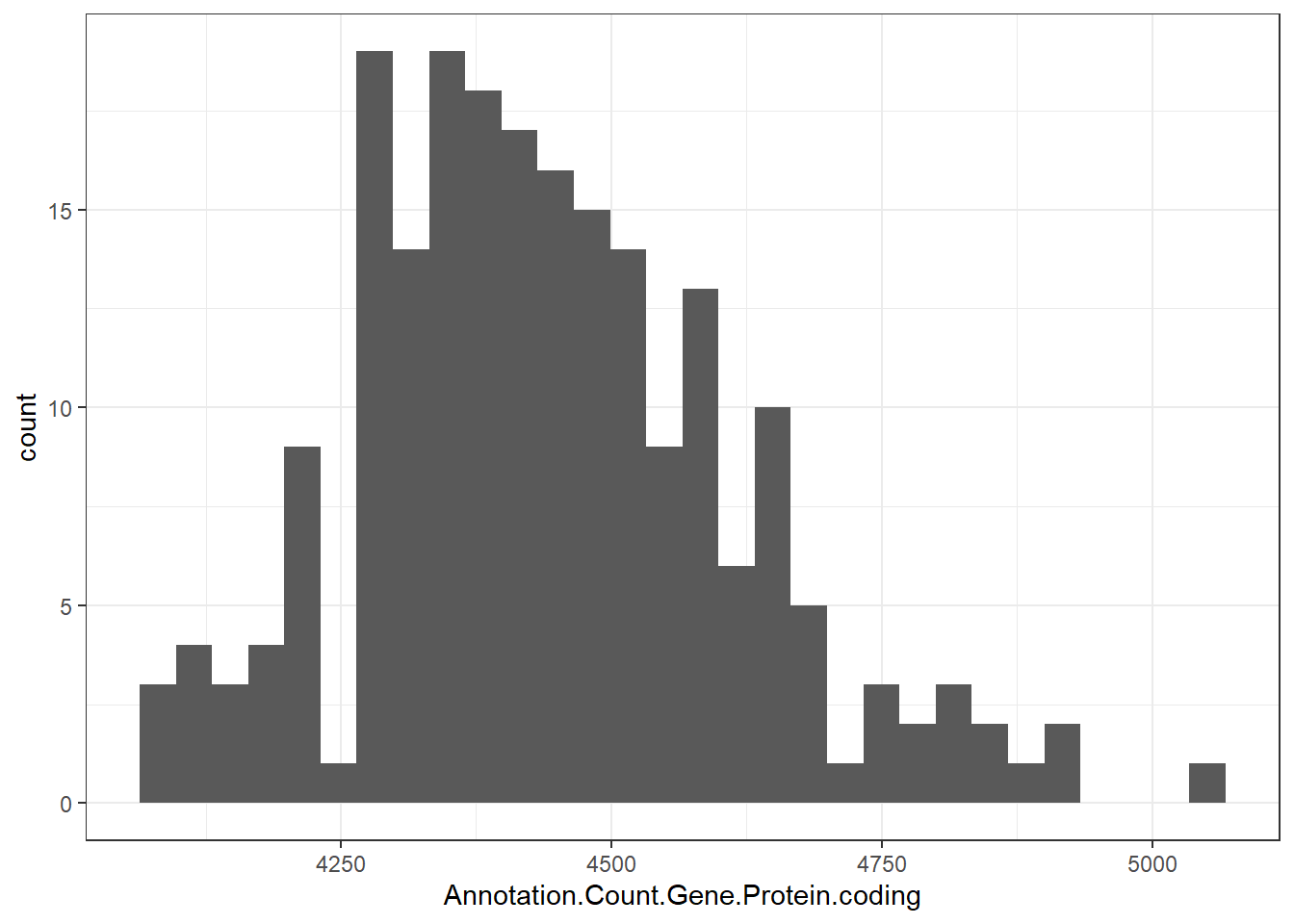
"Annotation.Count.Gene.Protein.coding" = genomes0$Annotation.Count.Gene.Protein.coding,
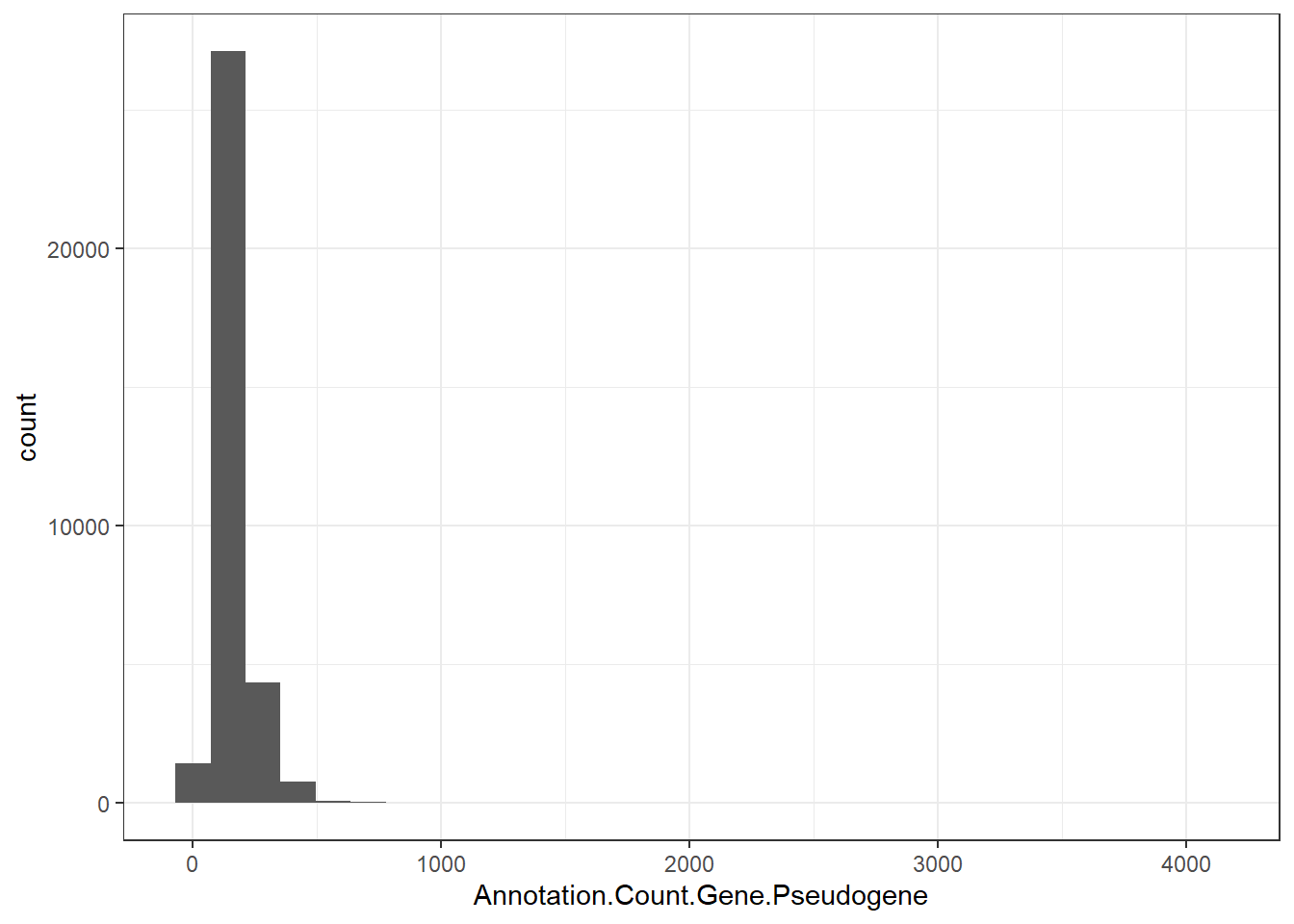
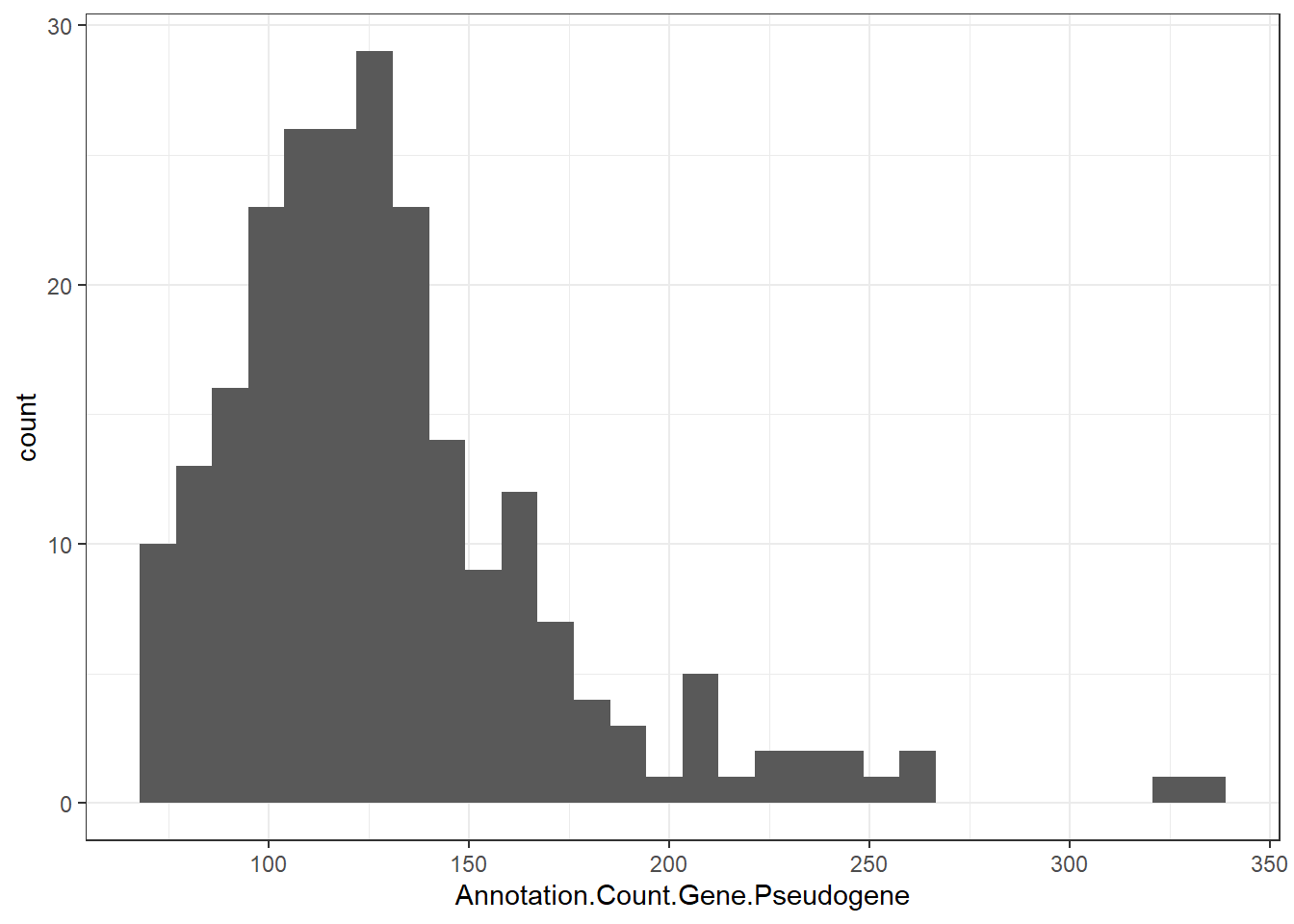
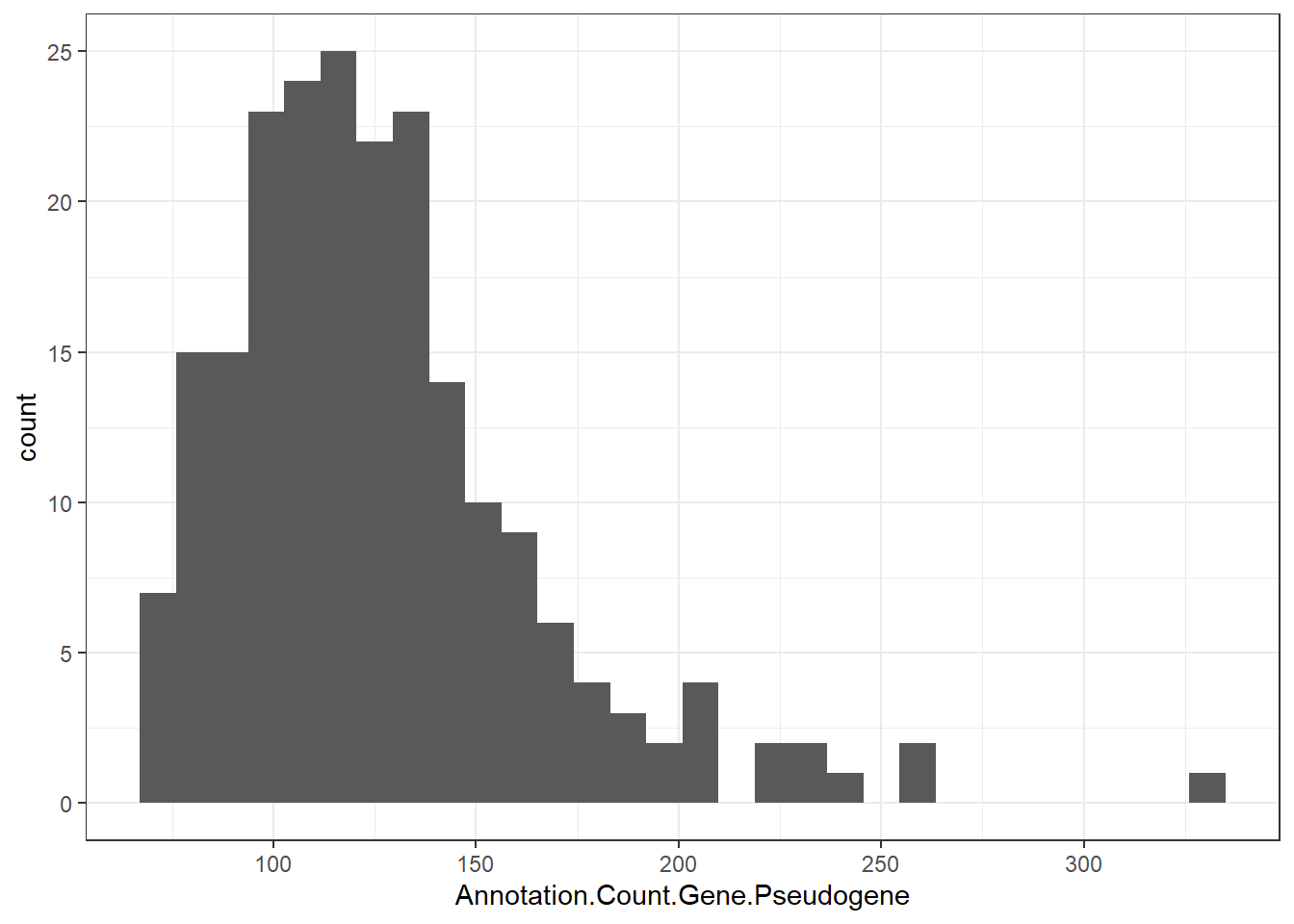
"Annotation.Count.Gene.Pseudogene" = genomes0$Annotation.Count.Gene.Pseudogene,
"genus" = genomes0$genus
)
genomes1 <- genomes1[(genomes1$genus %in% "Salmonella"), ]
genomes1 <- na.omit(genomes1)
write_csv(genomes1, here("data/processed-data/", "genomes1.csv"))