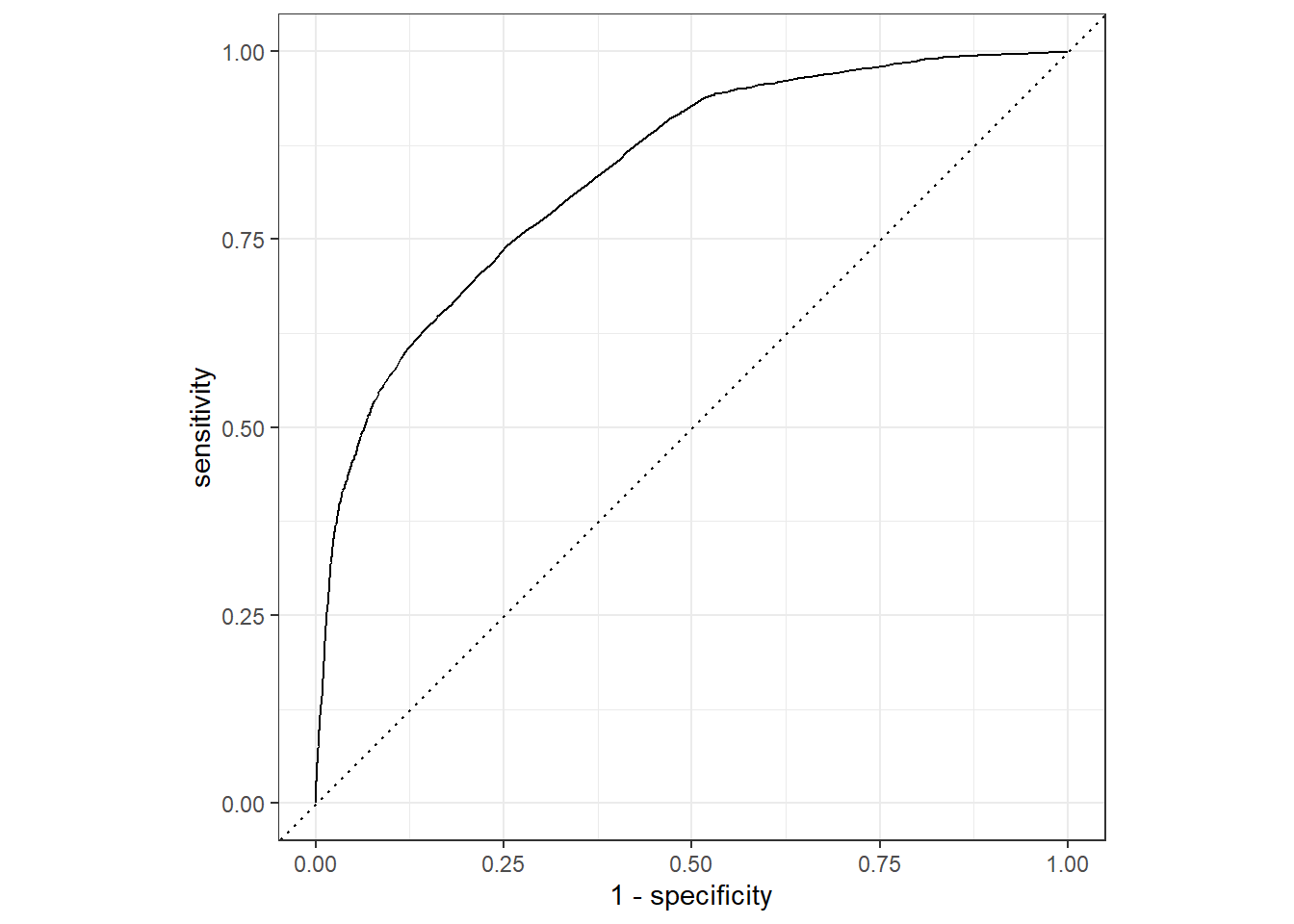
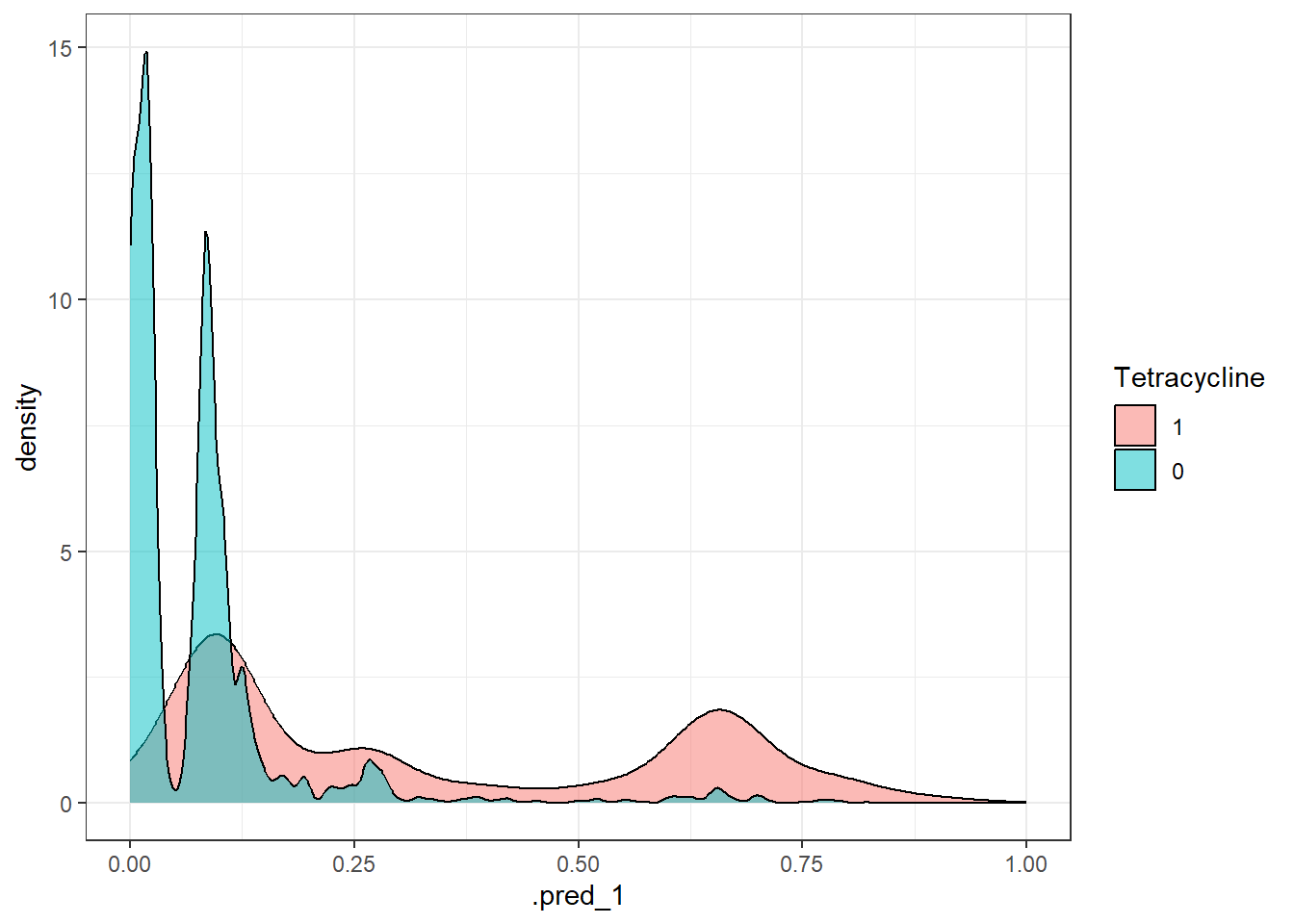
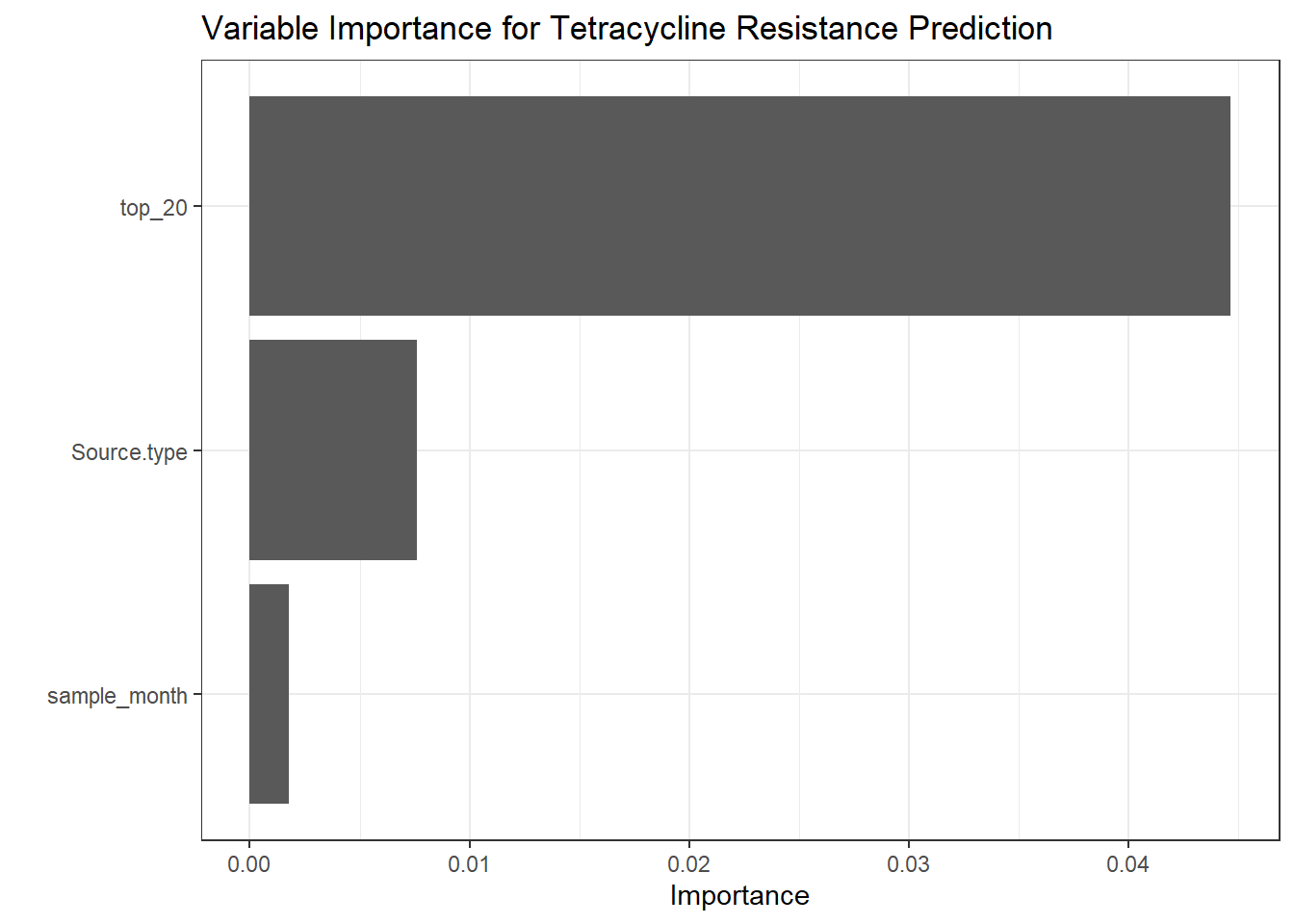
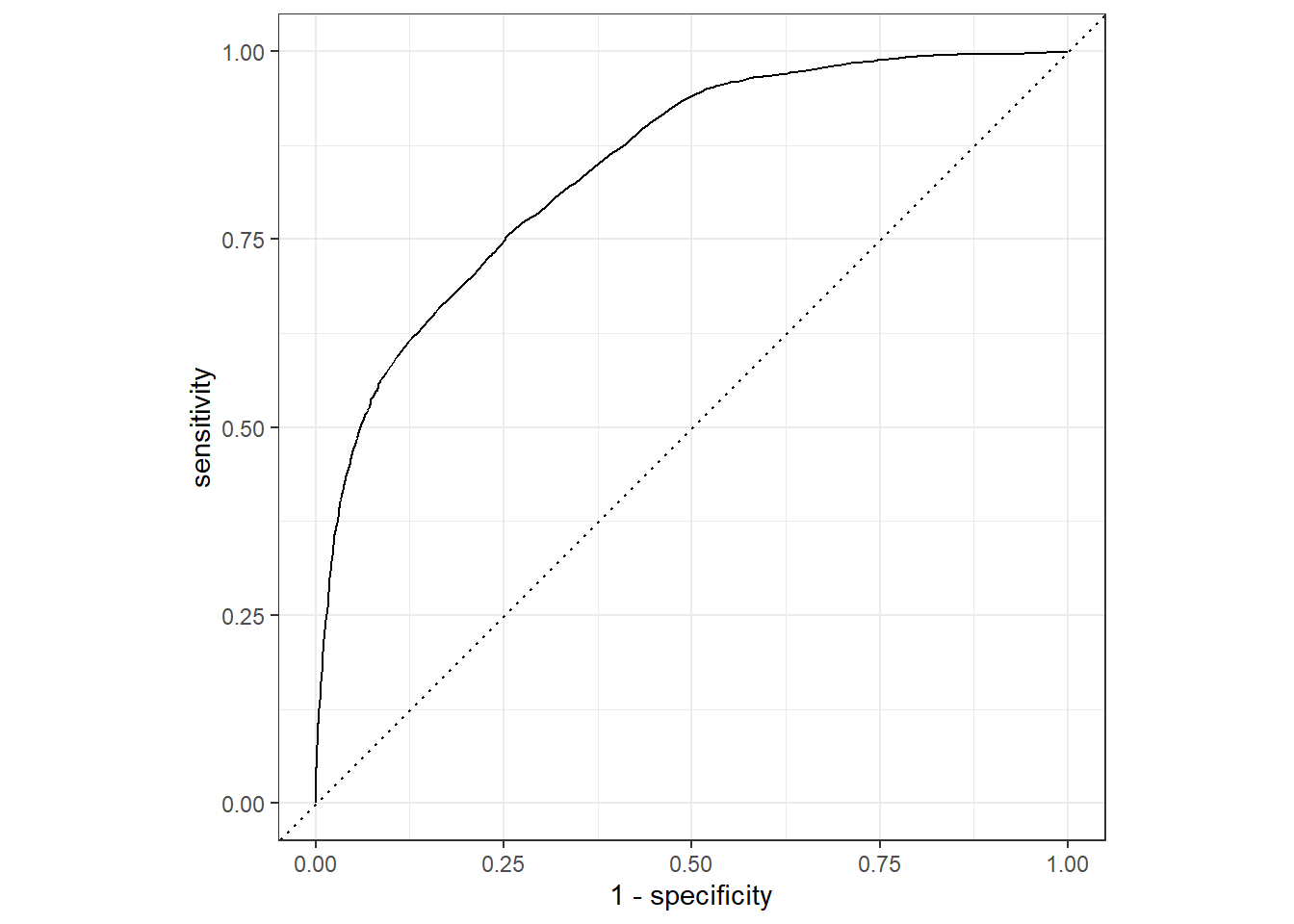
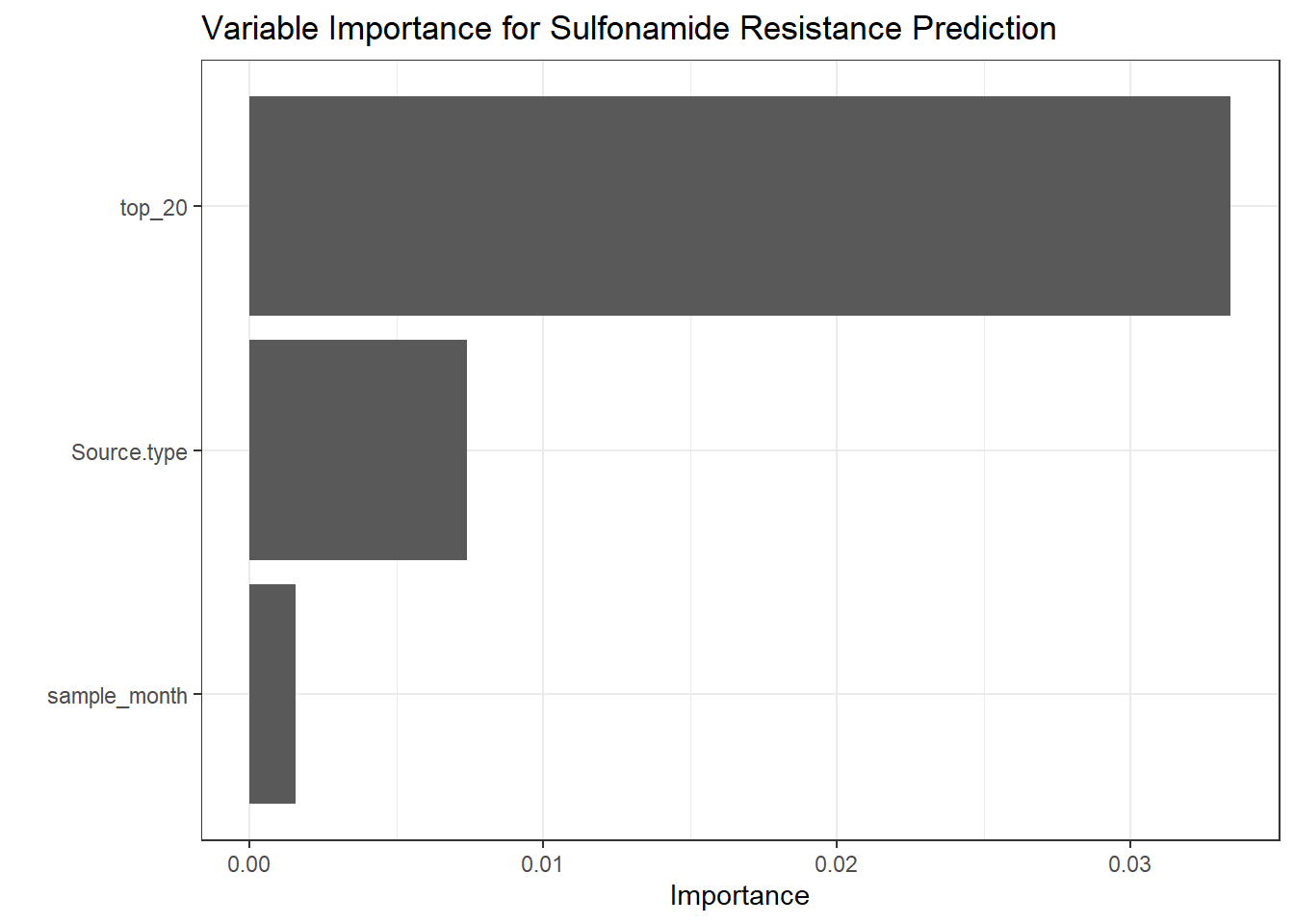
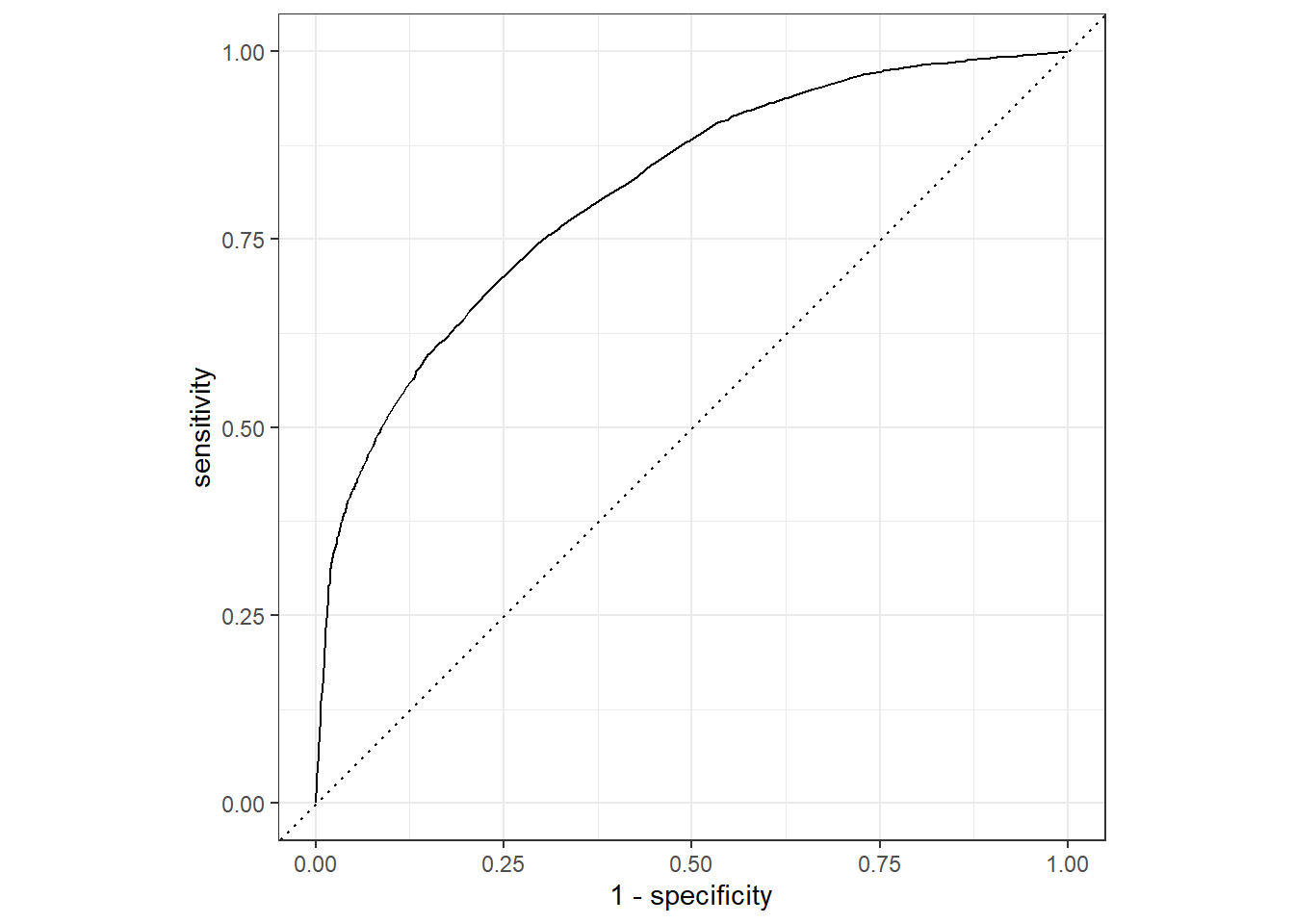
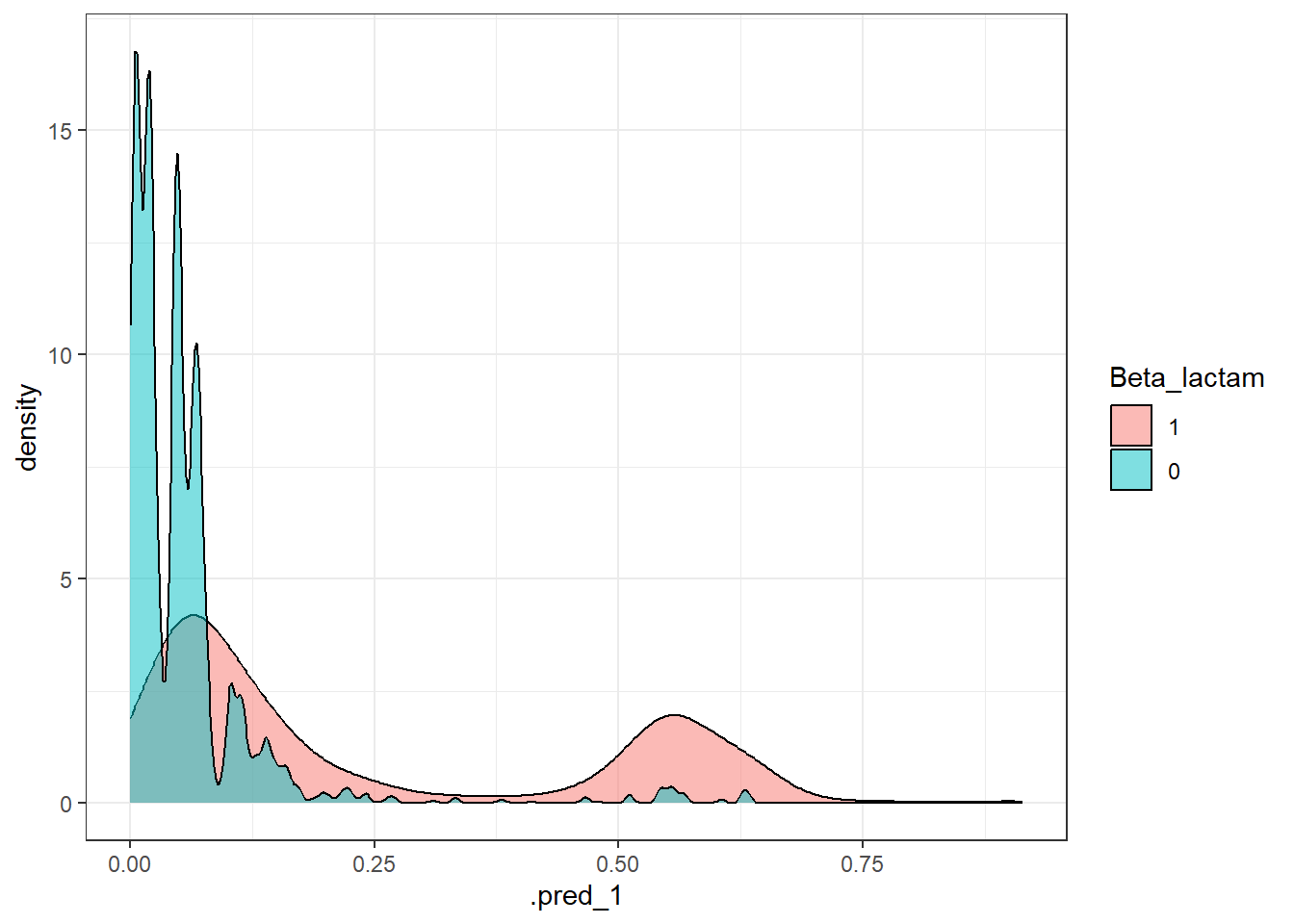
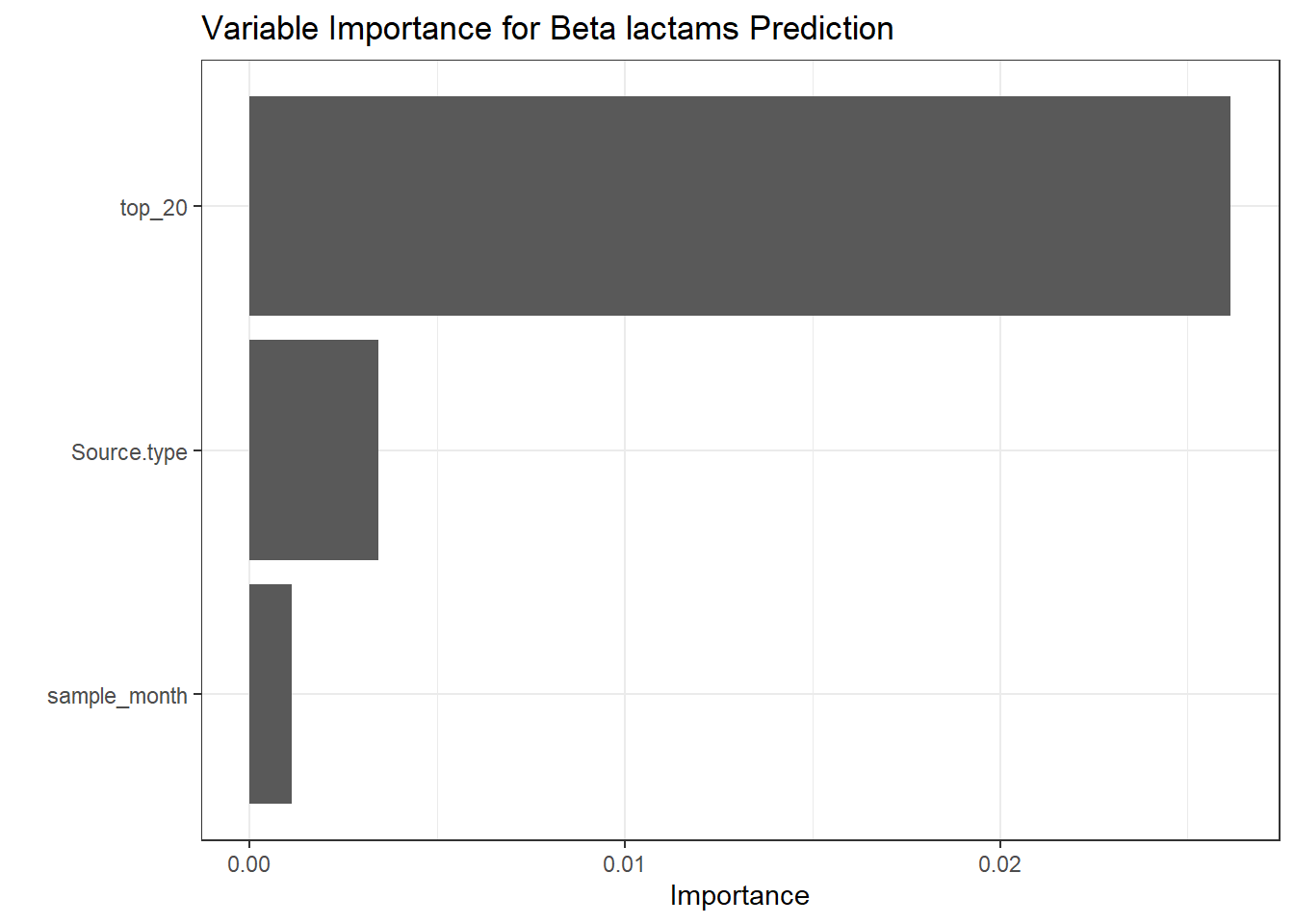
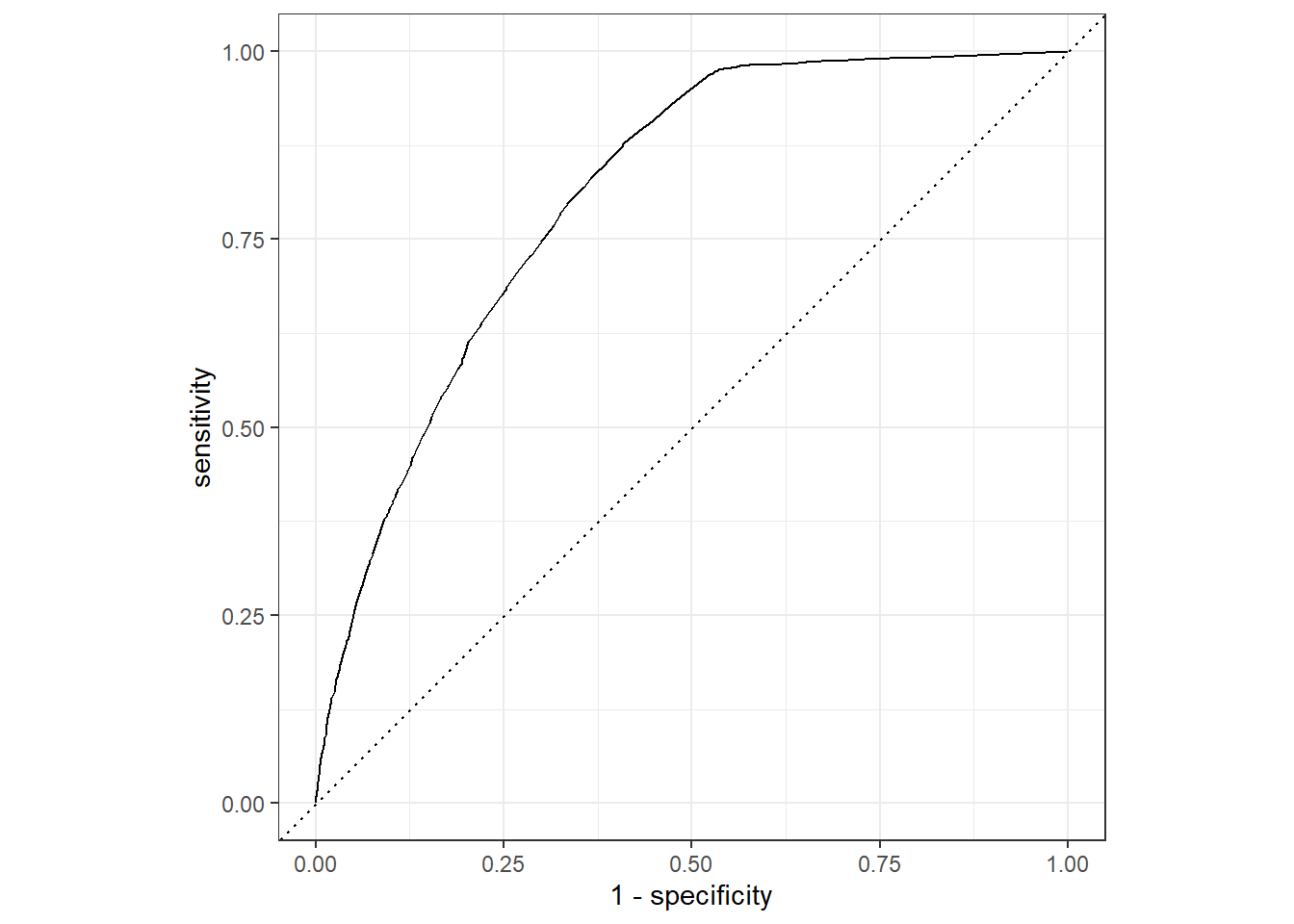
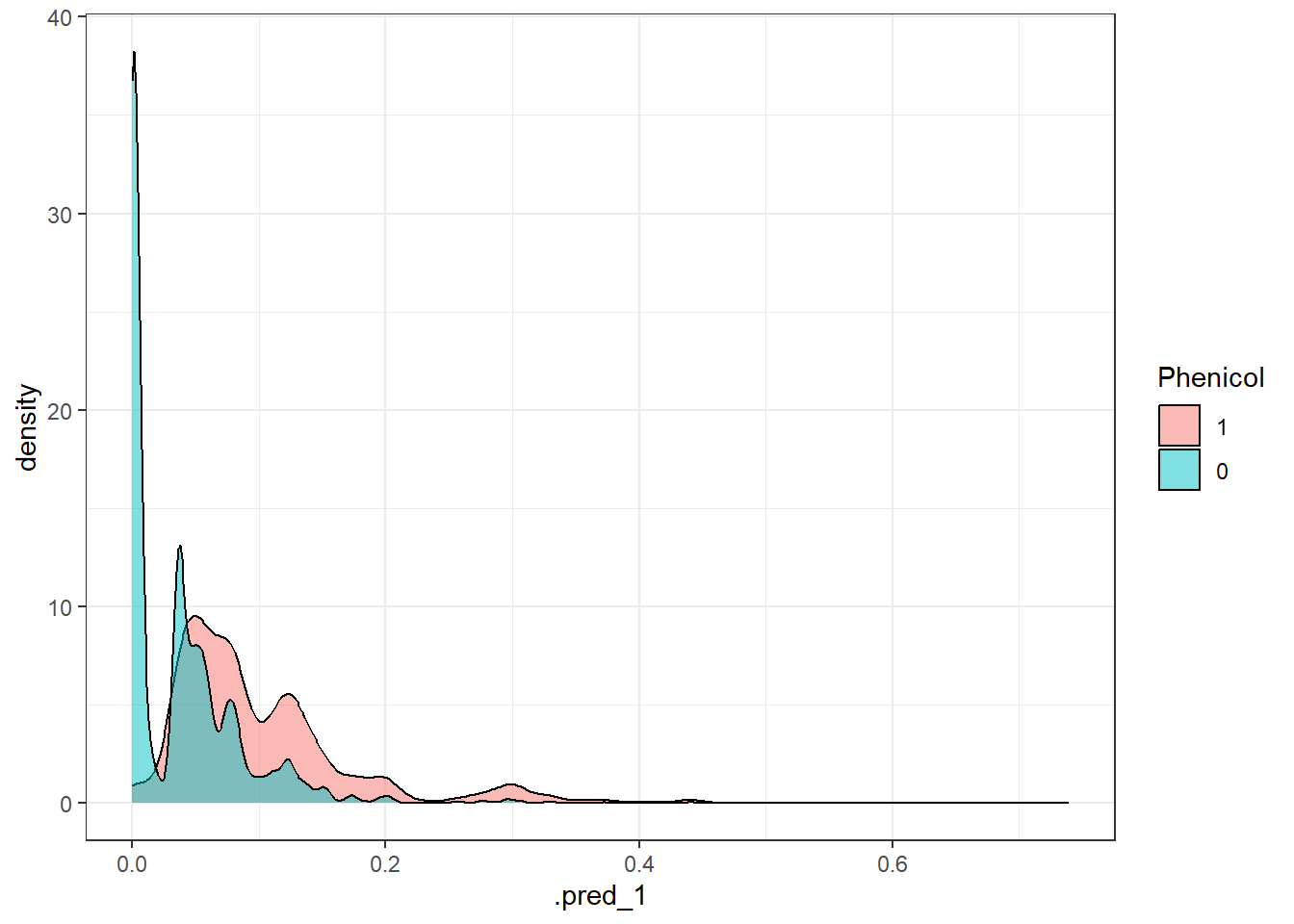
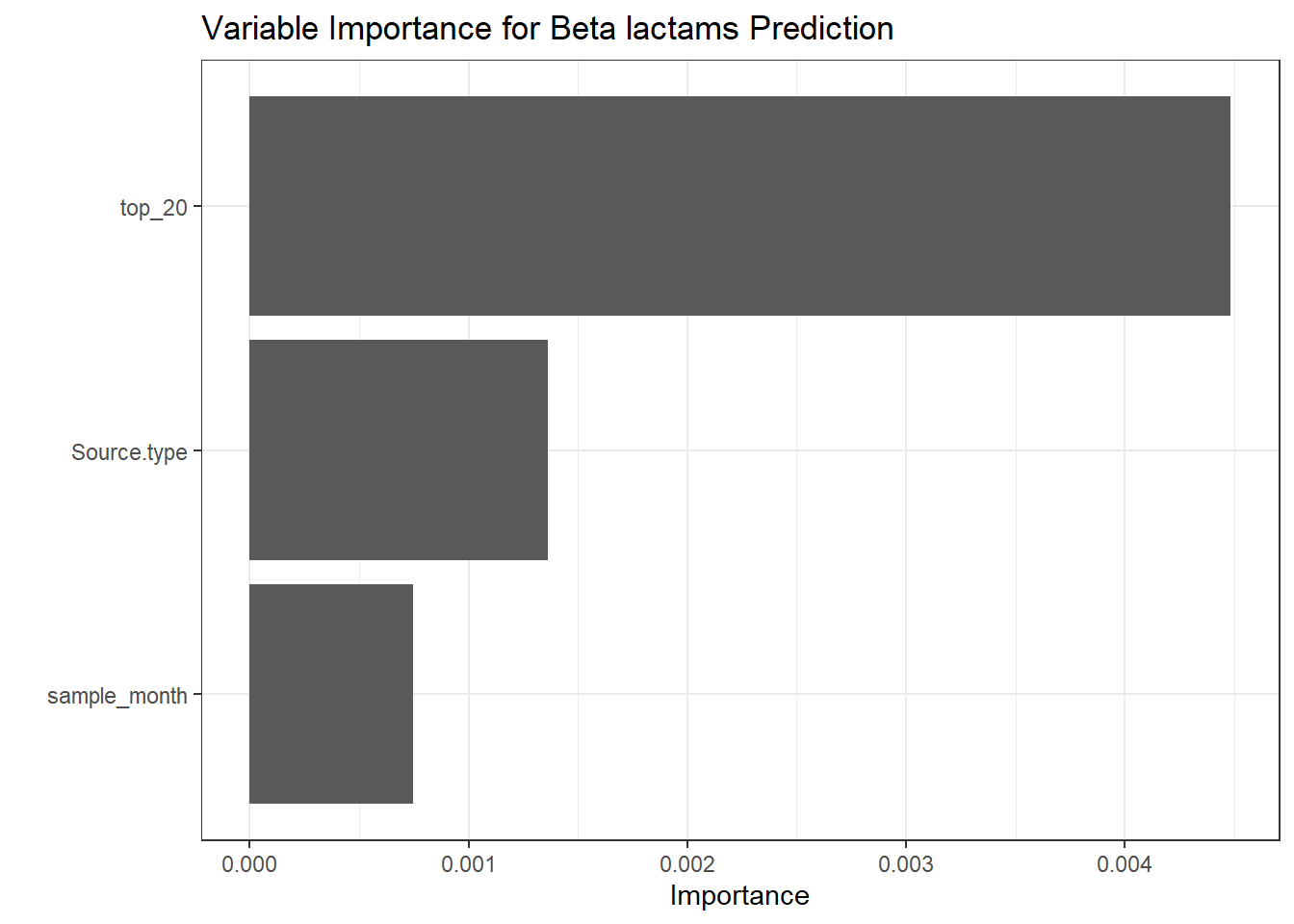
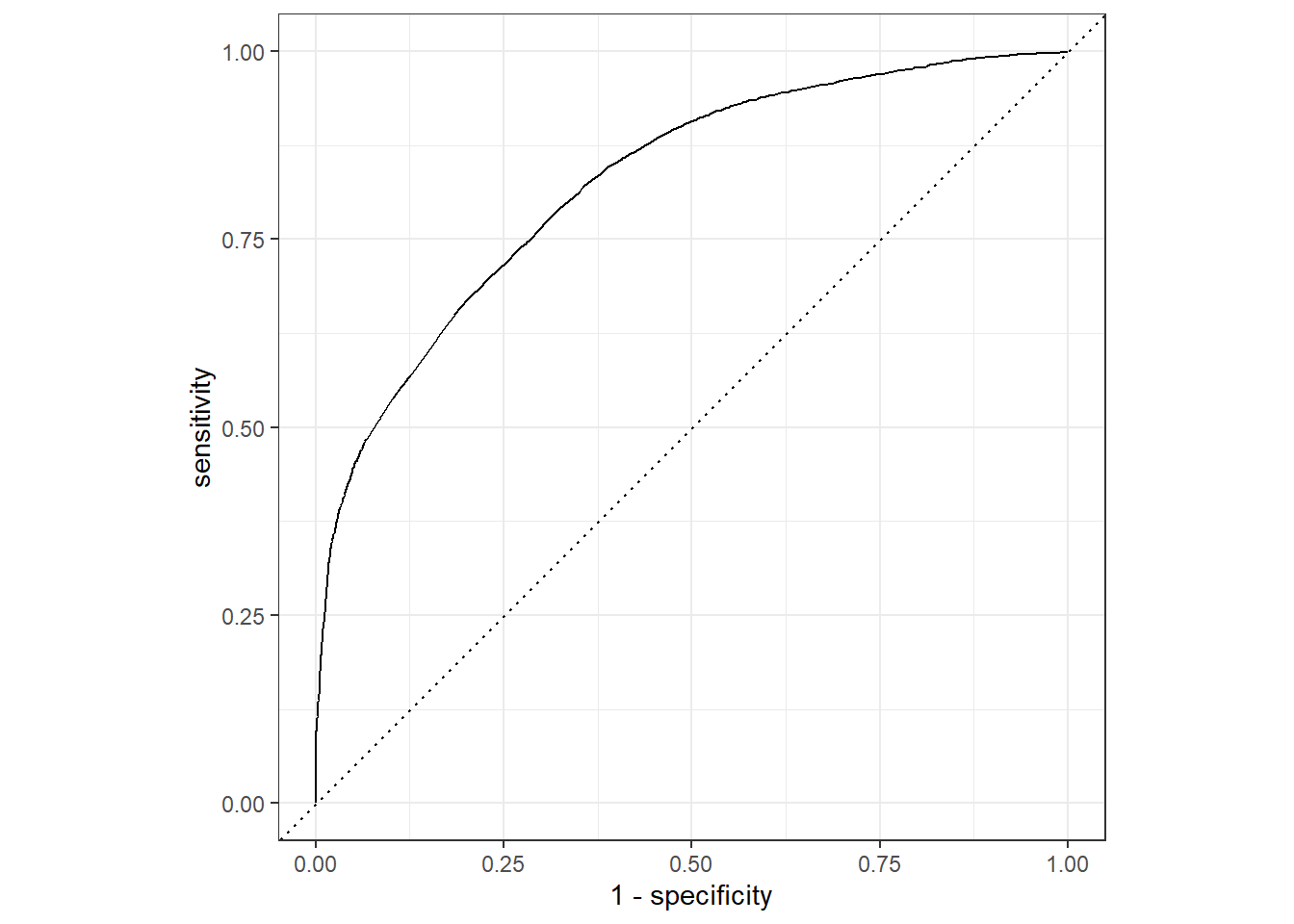
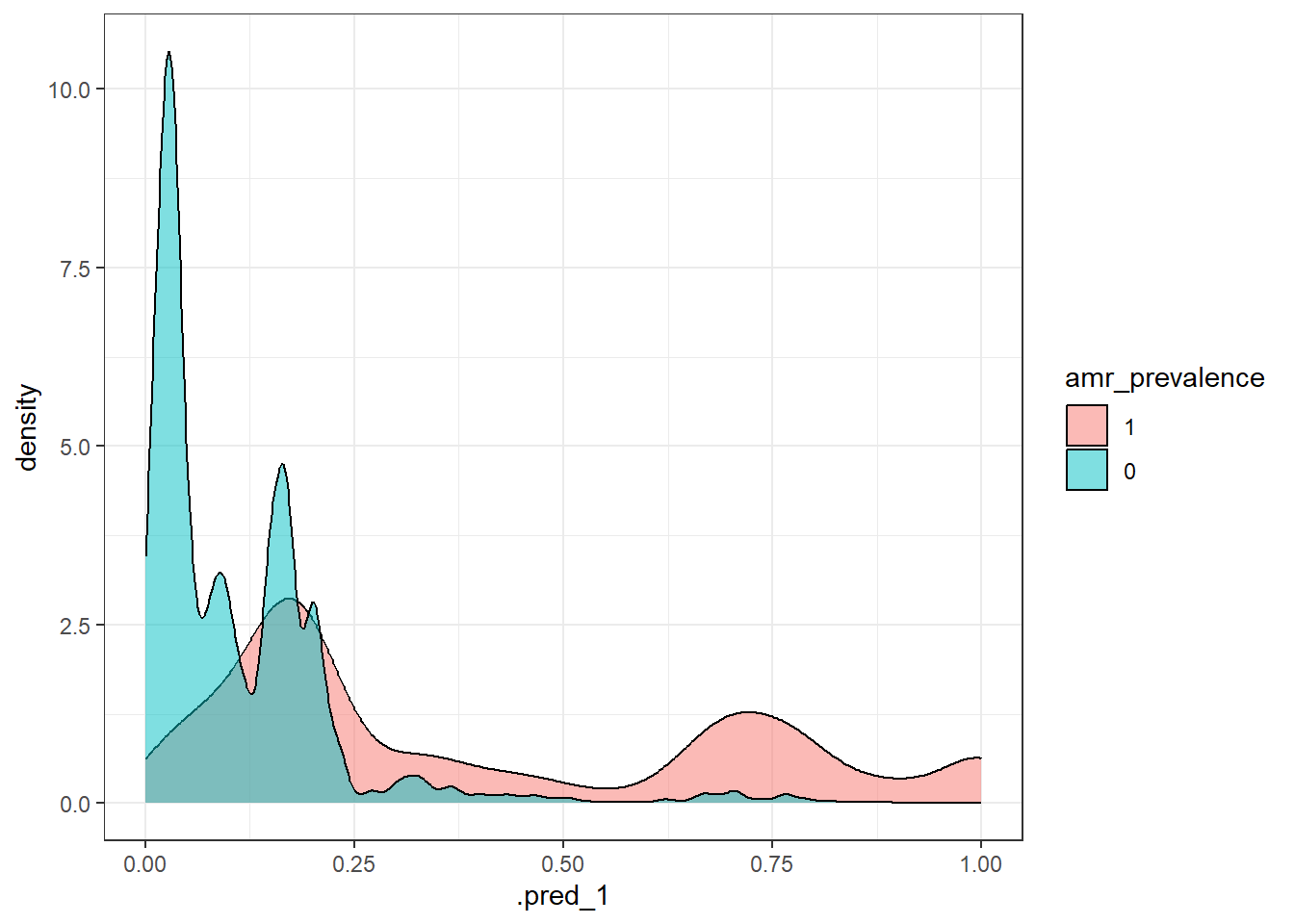
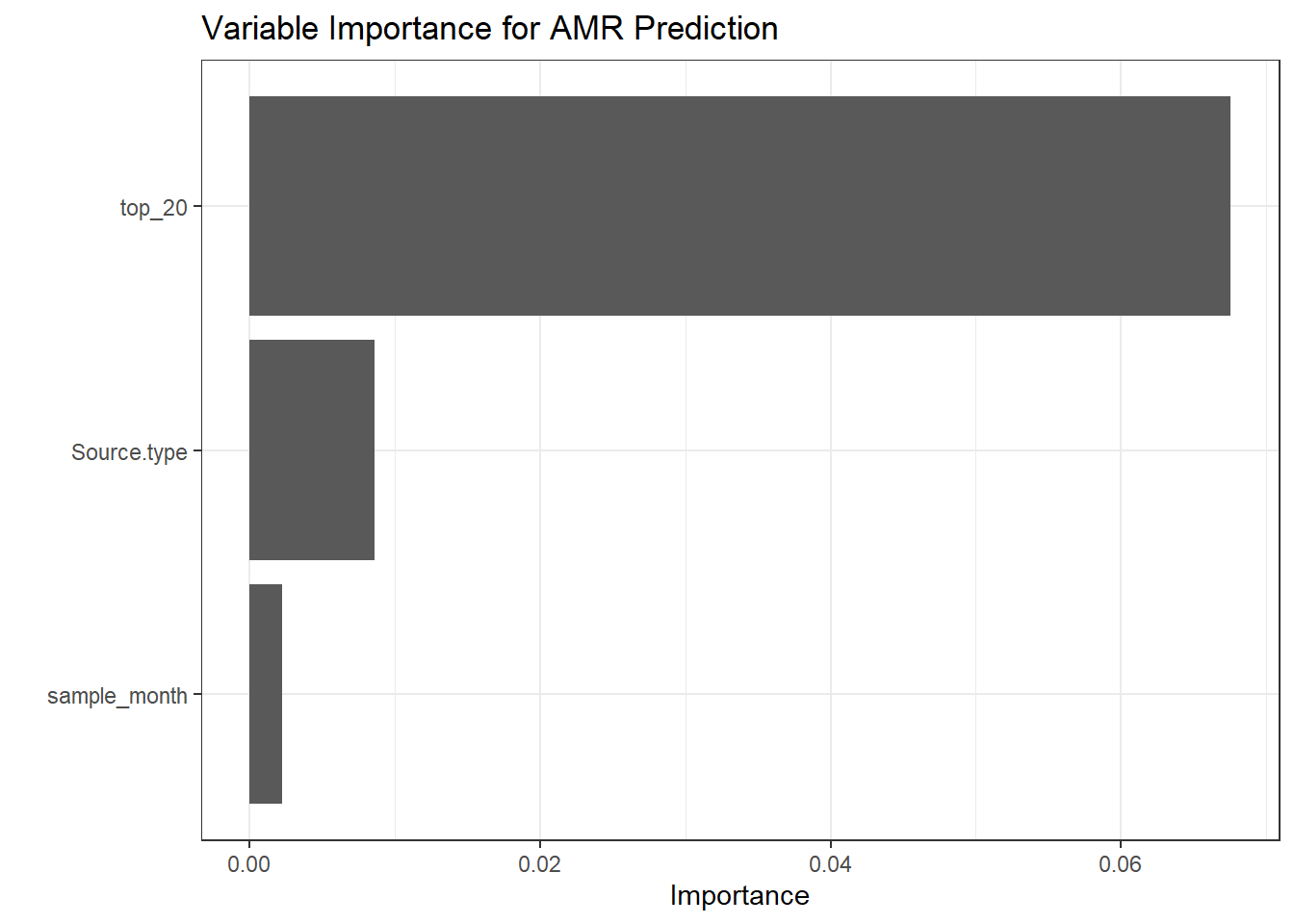
# Create a function to extract metrics from each prediction object
get_metrics <- function(pred_df, truth_col, model_name) {
list(
sens(pred_df, truth = !!sym(truth_col), estimate = .pred_class),
spec(pred_df, truth = !!sym(truth_col), estimate = .pred_class),
accuracy(pred_df, truth = !!sym(truth_col), estimate = .pred_class),
precision(pred_df, truth = !!sym(truth_col), estimate = .pred_class),
roc_auc(pred_df, truth = !!sym(truth_col), .pred_1)
) %>%
map_df(bind_rows) %>%
select(.metric, .estimate) %>%
pivot_wider(names_from = .metric, values_from = .estimate) %>%
mutate(Model = model_name)
}
# Combine all model results
all_results <- bind_rows(
get_metrics(test_pred_tet, "Tetracycline", "Tetracycline"),
get_metrics(test_pred_ami, "Aminoglycoside", "Aminoglycoside"),
get_metrics(test_pred_sul, "Sulfonamide", "Sulfonamide"),
get_metrics(test_pred_bet, "Beta_lactam", "Beta-lactam"),
get_metrics(test_pred_phe, "Phenicol", "Phenicol"),
get_metrics(test_pred5, "amr_prevalence", "General AMR")
) %>%
select(Model, roc_auc, sens, spec, accuracy, precision)
# Create the GT Table
all_results %>%
gt() %>%
tab_header(
title = "Random Forest Model Performance",
subtitle = "Predicting AMR Classes from Metadata"
) %>%
fmt_number(columns = 2:6, decimals = 3) %>%
cols_label(
Model = "Resistance Class",
roc_auc = "ROC AUC",
sens = "Sensitivity",
spec = "Specificity",
accuracy = "Accuracy",
precision = "Precision"
) %>%
data_color(columns = roc_auc, palette = "Blues") %>%
gtsave(here("results/tables/amr_model_performance.html"))